
Fabricante de medicamentos: Biogen Inc. (Updated: 2024-03-14)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
TECFIDERA® (dimetilfumarato) cápsulas de liberación retardada, para administración oral
Aprobación inicial en EE. UU.: 2013
CAMBIOS IMPORTANTES RECIENTES
| Advertencias y precauciones, reacciones gastrointestinales graves (5.7) | 12/2023 |
INDICACIONES Y USO
TECFIDERA está indicado para el tratamiento de las formas recurrentes de esclerosis múltiple (EM), incluidos el síndrome clínicamente aislado, la enfermedad recurrente-remitente y la enfermedad progresiva secundaria activa, en adultos. (1)
POSOLOGÍA Y ADMINISTRACIÓN
- Dosis inicial: 120 mg dos veces al día, por vía oral, durante 7 días (2.1)
- Dosis de mantenimiento después de 7 días: 240 mg dos veces al día, por vía oral (2.1)
- Trague las cápsulas de TECFIDERA enteras e intactas. No triture, mastique ni espolvoree el contenido de la cápsula sobre los alimentos (2.1)
- Tome TECFIDERA con o sin alimentos (2.1)
FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Cápsulas de liberación retardada: 120 mg y 240 mg (3)
CONTRAINDICACIONES
Hipersensibilidad conocida al dimetilfumarato o a cualquiera de los excipientes de TECFIDERA. (4)
ADVERTENCIAS Y PRECAUCIONES
- Anafilaxia y angioedema: Suspenda y no reinicie TECFIDERA si se producen estos síntomas. (5.1)
- Leucoencefalopatía multifocal progresiva (LMP): Suspenda TECFIDERA ante el primer signo o síntoma que sugiera LMP. (5.2)
- Herpes zóster y otras infecciones oportunistas graves: Considere la posibilidad de suspender TECFIDERA en casos de infección grave hasta que ésta se haya resuelto. (5.3)
- Linfopenia: Obtenga un hemograma completo, incluido el recuento de linfocitos, antes de iniciar TECFIDERA, después de 6 meses y cada 6 a 12 meses a partir de entonces. Considere la posibilidad de interrumpir TECFIDERA si los recuentos de linfocitos <0,5 x 109/L persisten durante más de seis meses. (5.4)
- Daño hepático: Obtenga los niveles de aminotransferasa sérica, fosfatasa alcalina y bilirrubina total antes de iniciar TECFIDERA y durante el tratamiento, según esté clínicamente indicado. Suspenda TECFIDERA si se sospecha daño hepático clínicamente significativo inducido por TECFIDERA. (5.5)
REACCIONES ADVERSAS
Las reacciones adversas más frecuentes (incidencia ≥10% y ≥2% placebo) fueron sofocos, dolor abdominal, diarrea y náuseas. (6.1)
Para notificar REACCIONES ADVERSAS SOSPECHOSAS, póngase en contacto con Biogen en el teléfono 1-800-456-2255 o con la FDA en el 1-800-FDA-1088 o en www.fda.gov/medwatch.
Consulte el punto 17 para obtener INFORMACIÓN DE ASESORAMIENTO AL PACIENTE y el etiquetado para pacientes aprobado por la FDA.
Revisado: 3/2024
Tabla de Contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Información sobre la dosificación
2.2 Análisis de sangre antes de iniciar la terapia
3 FORMAS Y FUERZAS DE DOSIFICACIÓN
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Anafilaxia y angioedema
5.2 Leucoencefalopatía multifocal progresiva
5.3 Herpes zóster y otras infecciones oportunistas graves
5.4 Linfopenia
5.5 Lesión hepática
5.6 Enrojecimiento
5.7 Reacciones gastrointestinales graves
6 REACCIONES ADVERSAS
6.1 Experiencia en ensayos clínicos
6.2 Experiencia postcomercialización
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.4 Uso pediátrico
8.5 Uso geriátrico
10 SOBREDOSIS
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, mutagénesis, deterioro de la fertilidad
13.2 Toxicología y/o farmacología animal
14 ESTUDIOS CLÍNICOS
16 CÓMO SE SUMINISTRA/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no están enumeradas.
1 INDICACIONES Y USO
TECFIDERA está indicado para el tratamiento de las formas recurrentes de esclerosis múltiple (EM), incluidos el síndrome clínicamente aislado, la enfermedad remitente-recurrente y la enfermedad progresiva secundaria activa, en adultos.
2 DOSIS Y ADMINISTRACIÓN
2.1 Información sobre la dosificación
La dosis inicial de TECFIDERA es de 120 mg dos veces al día por vía oral. Después de 7 días, la dosis debe aumentarse a la dosis de mantenimiento de 240 mg dos veces al día por vía oral. Se pueden considerar reducciones temporales de la dosis a 120 mg dos veces al día para las personas que no toleran la dosis de mantenimiento. Dentro de las 4 semanas, se debe reanudar la dosis recomendada de 240 mg dos veces al día. Se debe considerar la interrupción de TECFIDERA para los pacientes que no pueden tolerar el regreso a la dosis de mantenimiento. La incidencia de rubor puede reducirse administrando TECFIDERA con alimentos. Alternativamente, la administración de aspirina no recubierta entérica (hasta una dosis de 325 mg) 30 minutos antes de la dosificación de TECFIDERA puede reducir la incidencia o gravedad del rubor [ver Farmacología clínica (12.3)].
TECFIDERA debe tragarse entera e intacta. TECFIDERA no debe triturarse ni masticarse, y el contenido de la cápsula no debe espolvorearse sobre los alimentos. TECFIDERA se puede tomar con o sin alimentos.
2.2 Análisis de sangre antes de iniciar el tratamiento
Obtenga un hemograma completo (CBC) que incluya el recuento de linfocitos antes de iniciar el tratamiento [ver Advertencias y precauciones (5.4)].
Obtenga los niveles de aminotransferasa sérica, fosfatasa alcalina y bilirrubina total antes del tratamiento con TECFIDERA [ver Advertencias y precauciones (5.5)].
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
TECFIDERA está disponible en cápsulas de gelatina dura de liberación retardada que contienen 120 mg o 240 mg de dimetilfumarato. Las cápsulas de 120 mg tienen una tapa verde y un cuerpo blanco, impresos con “BG-12 120 mg” en tinta negra en el cuerpo. Las cápsulas de 240 mg tienen una tapa verde y un cuerpo verde, impresos con “BG-12 240 mg” en tinta negra en el cuerpo.
4 CONTRAINDICACIONES
TECFIDERA está contraindicado en pacientes con hipersensibilidad conocida al dimetilfumarato o a alguno de los excipientes de TECFIDERA. Las reacciones han incluido anafilaxia y angioedema [ver Advertencias y precauciones (5.1)].
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Anafilaxia y Angioedema
TECFIDERA puede causar anafilaxia y angioedema después de la primera dosis o en cualquier momento durante el tratamiento. Los signos y síntomas han incluido dificultad para respirar, urticaria e hinchazón de la garganta y la lengua. Se debe instruir a los pacientes que suspendan TECFIDERA y busquen atención médica inmediata si experimentan signos y síntomas de anafilaxia o angioedema.
5.2 Leucoencefalopatía Multifocal Progresiva
Se ha producido leucoencefalopatía multifocal progresiva (PML) en pacientes con EM tratados con TECFIDERA. La PML es una infección viral oportunista del cerebro causada por el virus JC (JCV) que normalmente solo ocurre en pacientes inmunocomprometidos y que generalmente conduce a la muerte o discapacidad grave. Se produjo un caso fatal de PML en un paciente que recibió TECFIDERA durante 4 años mientras participaba en un ensayo clínico. Durante el ensayo clínico, el paciente experimentó linfopenia prolongada (conteo de linfocitos predominantemente <0.5×109/L durante 3.5 años) mientras tomaba TECFIDERA [ver Advertencias y precauciones (5.4)]. El paciente no tenía otras afecciones médicas sistémicas identificadas que resultaran en una función inmunitaria comprometida y no había sido tratado previamente con natalizumab, que tiene una asociación conocida con la PML. El paciente tampoco estaba tomando ningún medicamento inmunosupresor o inmunomodulador de forma concomitante.
La PML también se ha producido en el entorno de postcomercialización en presencia de linfopenia (<0.9×109/L). Si bien el papel de la linfopenia en estos casos es incierto, los casos de PML se han producido predominantemente en pacientes con recuentos de linfocitos <0.8×109/L que persisten durante más de 6 meses.
Al primer signo o síntoma sugestivo de PML, suspenda TECFIDERA y realice una evaluación diagnóstica adecuada. Los síntomas típicos asociados con la PML son diversos, progresan de días a semanas e incluyen debilidad progresiva en un lado del cuerpo o torpeza de las extremidades, alteración de la visión y cambios en el pensamiento, la memoria y la orientación que conducen a confusión y cambios de personalidad.
Los hallazgos de la resonancia magnética pueden ser evidentes antes de los signos o síntomas clínicos. Se han reportado casos de PML, diagnosticados en base a los hallazgos de la resonancia magnética y la detección de ADN de JCV en el líquido cefalorraquídeo en ausencia de signos o síntomas clínicos específicos de la PML, en pacientes tratados con otros medicamentos para la EM asociados con la PML. Muchos de estos pacientes posteriormente se volvieron sintomáticos con PML. Por lo tanto, el monitoreo con resonancia magnética para detectar signos que puedan ser consistentes con la PML puede ser útil, y cualquier hallazgo sospechoso debe conducir a una investigación adicional para permitir un diagnóstico temprano de la PML, si está presente. Se ha informado una menor mortalidad y morbilidad relacionadas con la PML después de la interrupción de otro medicamento para la EM asociado con la PML en pacientes con PML que inicialmente eran asintomáticos en comparación con los pacientes con PML que tenían signos y síntomas clínicos característicos en el momento del diagnóstico. No se sabe si estas diferencias se deben a la detección temprana y la interrupción del tratamiento de la EM o a las diferencias en la enfermedad en estos pacientes.
5.3 Herpes Zoster y Otras Infecciones Oportunistas Graves
Se han producido casos graves de herpes zóster con TECFIDERA, incluido el herpes zóster diseminado, el herpes zóster oftálmico, el herpes zóster meningoencefalitis y el herpes zóster meningomielitis. Estos eventos pueden ocurrir en cualquier momento durante el tratamiento. Monitoree a los pacientes que toman TECFIDERA para detectar signos y síntomas de herpes zóster. Si se produce herpes zóster, se debe administrar el tratamiento adecuado para el herpes zóster.
Se han producido otras infecciones oportunistas graves con TECFIDERA, incluidos casos de infecciones virales graves (virus del herpes simple, virus del Nilo Occidental, citomegalovirus), fúngicas (Candida y Aspergillus) y bacterianas (Nocardia, Listeria monocytogenes, Mycobacterium tuberculosis). Estas infecciones se han reportado en pacientes con recuentos de linfocitos absolutos (ALC) reducidos, así como en pacientes con ALC normales. Estas infecciones han afectado el cerebro, las meninges, la médula espinal, el tracto gastrointestinal, los pulmones, la piel, los ojos y los oídos. Los pacientes con síntomas y signos consistentes con cualquiera de estas infecciones deben someterse a una evaluación diagnóstica inmediata y recibir el tratamiento adecuado.
Considere suspender el tratamiento con TECFIDERA en pacientes con herpes zóster u otras infecciones graves hasta que la infección se haya resuelto [ver Reacciones adversas (6.2)].
5.4 Linfopenia
TECFIDERA puede disminuir el conteo de linfocitos. En los ensayos controlados con placebo para la EM, el conteo medio de linfocitos disminuyó aproximadamente un 30% durante el primer año de tratamiento con TECFIDERA y luego se mantuvo estable. Cuatro semanas después de suspender TECFIDERA, el conteo medio de linfocitos aumentó pero no volvió a la línea de base. El seis por ciento (6%) de los pacientes con TECFIDERA y <1% de los pacientes con placebo experimentaron conteos de linfocitos <0.5×109/L (límite inferior de lo normal 0.91×109/L). La incidencia de infecciones (60% vs 58%) e infecciones graves (2% vs 2%) fue similar en pacientes tratados con TECFIDERA o placebo, respectivamente. No se observó un aumento en la incidencia de infecciones graves en pacientes con conteos de linfocitos <0.8×109/L o ≤0.5×109/L en ensayos controlados, aunque un paciente en un estudio de extensión desarrolló PML en el contexto de linfopenia prolongada (conteos de linfocitos predominantemente <0.5×109/L durante 3.5 años) [ver Advertencias y precauciones (5.2)].
En ensayos clínicos controlados y no controlados, el 2% de los pacientes experimentaron linfopenia prolongada y grave (definida como conteos de linfocitos <0.5 x 109/L durante al menos seis meses); en este grupo de pacientes, la mayoría de los conteos de linfocitos se mantuvieron <0.5×109/L con terapia continua. En estos pacientes con linfopenia prolongada y grave, la mediana del tiempo para que los conteos de linfocitos vuelvan a la normalidad después de suspender TECFIDERA fue de 96.0 semanas.
En estos estudios clínicos controlados y no controlados, entre los pacientes que no experimentaron linfopenia prolongada y grave durante el tratamiento, las medianas del tiempo para que los conteos de linfocitos vuelvan a la normalidad después de suspender TECFIDERA fueron las siguientes:
- 4.3 semanas en pacientes con linfopenia leve (recuento de linfocitos ≥0.8×109/L) al suspender el tratamiento,
- 10.0 semanas en pacientes con linfopenia moderada (recuento de linfocitos de 0.5 a <0.8×109/L) al suspender el tratamiento, y
- 16.7 semanas en pacientes con linfopenia grave (recuento de linfocitos <0.5×109/L) al suspender el tratamiento.
TECFIDERA no se ha estudiado en pacientes con recuentos bajos de linfocitos preexistentes.
Obtenga un hemograma completo, incluido el recuento de linfocitos, antes de iniciar el tratamiento con TECFIDERA, 6 meses después de comenzar el tratamiento y luego cada 6 a 12 meses después, y según esté clínicamente indicado. Considere la interrupción de TECFIDERA en pacientes con recuentos de linfocitos inferiores a 0.5 x 109/L que persistan durante más de seis meses. Dada la posibilidad de recuperación tardía de los recuentos de linfocitos, continúe obteniendo recuentos de linfocitos hasta que se recuperen si se suspende o interrumpe TECFIDERA debido a linfopenia. Considere la posibilidad de suspender el tratamiento en pacientes con infecciones graves hasta que se resuelvan. Las decisiones sobre si reiniciar o no TECFIDERA deben individualizarse en función de las circunstancias clínicas.
5.5 Lesión hepática
Se han notificado casos clínicamente significativos de lesión hepática en pacientes tratados con TECFIDERA en el contexto de la poscomercialización. El inicio ha variado de unos pocos días a varios meses después del inicio del tratamiento con TECFIDERA. Se han observado signos y síntomas de lesión hepática, incluida la elevación de las aminotransferasas séricas a más de 5 veces el límite superior de lo normal y la elevación de la bilirrubina total a más de 2 veces el límite superior de lo normal. Estas anomalías se resolvieron al suspender el tratamiento. Algunos casos requirieron hospitalización. Ninguno de los casos notificados provocó insuficiencia hepática, trasplante de hígado o muerte. Sin embargo, la combinación de nuevas elevaciones de las aminotransferasas séricas con niveles elevados de bilirrubina causados por lesión hepatocelular inducida por fármacos es un predictor importante de lesión hepática grave que puede provocar insuficiencia hepática aguda, trasplante de hígado o muerte en algunos pacientes.
Se observaron elevaciones de las transaminasas hepáticas (la mayoría no superiores a 3 veces el límite superior de lo normal) durante los ensayos controlados [ver Reacciones adversas (6.1)].
Obtenga los niveles de aminotransferasas séricas, fosfatasa alcalina (ALP) y bilirrubina total antes del tratamiento con TECFIDERA y durante el tratamiento, según esté clínicamente indicado. Suspenda TECFIDERA si se sospecha una lesión hepática clínicamente significativa inducida por TECFIDERA.
5.6 Rubor
TECFIDERA puede causar rubor (por ejemplo, calor, enrojecimiento, picazón y/o sensación de ardor). En los ensayos clínicos, el 40% de los pacientes tratados con TECFIDERA experimentaron rubor. Los síntomas de rubor generalmente comenzaron poco después de iniciar TECFIDERA y generalmente mejoraron o se resolvieron con el tiempo. En la mayoría de los pacientes que experimentaron rubor, fue de intensidad leve o moderada. El tres por ciento (3%) de los pacientes suspendieron TECFIDERA por rubor y <1% tuvo síntomas graves de rubor que no fueron potencialmente mortales pero que llevaron a la hospitalización. La administración de TECFIDERA con alimentos puede reducir la incidencia de rubor. Alternativamente, la administración de aspirina no recubierta entérica (hasta una dosis de 325 mg) 30 minutos antes de la dosificación de TECFIDERA puede reducir la incidencia o gravedad del rubor [ver Dosificación y administración (2.1) y Farmacología clínica (12.3)].
5.7 Reacciones gastrointestinales graves
Se han notificado reacciones gastrointestinales graves, incluida la perforación, la ulceración, la hemorragia y la obstrucción, algunas con desenlaces fatales, en el contexto de la poscomercialización con el uso de ésteres de ácido fumárico, incluido TECFIDERA, con o sin uso concomitante de aspirina. La mayoría de estos eventos han ocurrido dentro de los 6 meses posteriores al inicio del tratamiento con ésteres de ácido fumárico. En los ensayos clínicos controlados, la incidencia de eventos adversos gastrointestinales graves fue del 1% en pacientes tratados con TECFIDERA; estos eventos, ninguno de los cuales fue fatal, incluyeron vómitos (0.3%) y dolor abdominal (0.3%) [ver Reacciones adversas (6.1)].
Controle a los pacientes, evalúe con prontitud y suspenda TECFIDERA por signos y síntomas gastrointestinales graves nuevos o que empeoren.
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas importantes se describen en otras partes de la etiqueta:
- Anafilaxia y Angioedema [ver Advertencias y precauciones (5.1)].
- Leucoencefalopatía multifocal progresiva [ver Advertencias y precauciones (5.2)].
- Herpes Zoster y otras infecciones oportunistas graves [ver Advertencias y precauciones (5.3)].
- Linfopenia [ver Advertencias y precauciones (5.4)].
- Lesión hepática [ver Advertencias y precauciones (5.5)].
- Rubor [ver Advertencias y precauciones (5.6)].
- Reacciones gastrointestinales graves [ver Advertencias y precauciones (5.7)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica clínica.
En estudios clínicos controlados con placebo y no controlados, un total de 2513 pacientes han recibido TECFIDERA y han sido seguidos durante períodos de hasta 13 años con una exposición general de 11,318 personas-año. Aproximadamente 1169 pacientes han recibido más de 5 años de tratamiento con TECFIDERA, y 426 pacientes han recibido al menos 10 años de tratamiento con TECFIDERA.
Reacciones adversas en ensayos controlados con placebo
En los dos estudios bien controlados que demostraron eficacia, 1529 pacientes recibieron TECFIDERA con una exposición general de 2244 personas-año [ver Estudios clínicos (14)].
Las reacciones adversas que se presentan en la tabla a continuación se basan en información de seguridad de 769 pacientes tratados con TECFIDERA 240 mg dos veces al día y 771 pacientes tratados con placebo.
Las reacciones adversas más comunes (incidencia ≥10% y ≥2% más que placebo) para TECFIDERA fueron rubor, dolor abdominal, diarrea y náuseas.
| TECFIDERA N=769 % |
Placebo N=771 % |
|
| Rubor | 40 | 6 |
| Dolor abdominal | 18 | 10 |
| Diarrea | 14 | 11 |
| Náuseas | 12 | 9 |
| Vómitos | 9 | 5 |
| Prurito | 8 | 4 |
| Erupción | 8 | 3 |
| Albúmina en orina presente | 6 | 4 |
| Eritema | 5 | 1 |
| Dispepsia | 5 | 3 |
| Aspartato aminotransferasa aumentada | 4 | 2 |
| Linfopenia | 2 | <1 |
Gastrointestinal
TECFIDERA causó eventos GI (por ejemplo, náuseas, vómitos, diarrea, dolor abdominal y dispepsia). La incidencia de eventos GI fue mayor al inicio del tratamiento (principalmente en el mes 1) y generalmente disminuyó con el tiempo en los pacientes tratados con TECFIDERA en comparación con el placebo. El cuatro por ciento (4%) de los pacientes tratados con TECFIDERA y menos del 1% de los pacientes con placebo interrumpieron el tratamiento debido a eventos gastrointestinales. La incidencia de eventos GI graves fue del 1% en los pacientes de ensayos clínicos tratados con TECFIDERA; estos eventos, ninguno de los cuales fue fatal, incluyeron vómitos (0,3%) y dolor abdominal (0,3%).
Hepatic Transaminases
Se observó un aumento de la incidencia de elevaciones de las transaminasas hepáticas en pacientes tratados con TECFIDERA, principalmente durante los primeros seis meses de tratamiento, y la mayoría de los pacientes con elevaciones tuvieron niveles < 3 veces el límite superior normal (ULN) durante los ensayos controlados. Las elevaciones de la alanina aminotransferasa y la aspartato aminotransferasa a ≥ 3 veces el ULN ocurrieron en un pequeño número de pacientes tratados tanto con TECFIDERA como con placebo y se equilibraron entre los grupos. No hubo elevaciones en las transaminasas ≥ 3 veces el ULN con elevaciones concomitantes en la bilirrubina total > 2 veces el ULN. Las interrupciones debido a transaminasas hepáticas elevadas fueron < 1% y fueron similares en pacientes tratados con TECFIDERA o placebo.
6.2 Experiencia postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de TECFIDERA. Debido a que estas reacciones se informan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Trastornos gastrointestinales: Pancreatitis aguda; Perforación, ulceración, obstrucción y hemorragia gastrointestinal [ver Advertencias y precauciones (5.7)]
Trastornos hepatobiliares: Anormalidades de la función hepática (elevaciones en las transaminasas ≥ 3 veces ULN con elevaciones concomitantes en la bilirrubina total > 2 veces ULN) [ver Advertencias y precauciones (5.5)]
Infecciones e infestaciones: Infección por herpes zóster y otras infecciones oportunistas graves [ver Advertencias y precauciones (5.3)]
Trastornos respiratorios, torácicos y mediastínicos: Rinorrea
Piel y tejido subcutáneo: Alopecia
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de riesgos
Los datos disponibles del Registro de Embarazos de TECFIDERA, los estudios observacionales y la farmacovigilancia con el uso de dimetilfumarato en mujeres embarazadas no han indicado un mayor riesgo de defectos de nacimiento importantes, aborto espontáneo u otros resultados adversos maternos o fetales. La mayoría de las exposiciones notificadas al dimetilfumarato se produjeron durante el primer trimestre del embarazo (véase Datos). En animales, se observaron efectos adversos sobre la supervivencia, el crecimiento, la maduración sexual y la función neuroconductual de las crías cuando se administró dimetilfumarato (DMF) durante el embarazo y la lactancia a dosis clínicamente relevantes (véase Datos).
Se desconoce el riesgo de base de defectos de nacimiento importantes y aborto espontáneo para la población indicada. Todos los embarazos tienen un riesgo de base de defectos de nacimiento, pérdida u otros resultados adversos. En la población general de los Estados Unidos, el riesgo de base estimado de defectos de nacimiento importantes y aborto espontáneo en embarazos clínicamente reconocidos es del 2-4% y del 15-20%, respectivamente.
Datos
Datos humanos
En un Registro de Embarazos de TECFIDERA observacional prospectivo (2013-2022), la tasa de defectos de nacimiento importantes entre 362 nacidos vivos y mortinatos de mujeres que estuvieron expuestas al dimetilfumarato durante el embarazo fue del 3.6% (IC del 95%: 1.9-6.1). No se identificó ningún patrón específico de defectos de nacimiento importantes. Entre las limitaciones potenciales importantes del estudio se encuentran la clasificación errónea de la exposición, la falta de ajuste por factores de confusión y la falta de una cohorte de comparación interna.
Datos en animales
En ratas a las que se administró DMF por vía oral (25, 100, 250 mg/kg/día) durante la organogénesis, se observó toxicidad embriofetal (reducción del peso corporal fetal y retraso de la osificación) con la dosis más alta probada. Esta dosis también produjo evidencia de toxicidad materna (reducción del peso corporal). La exposición plasmática (AUC) para el monometilfumarato (MMF), el principal metabolito circulante, a la dosis sin efecto es aproximadamente tres veces mayor que en humanos a la dosis humana recomendada (DHR) de 480 mg/día. En conejos a los que se administró DMF por vía oral (25, 75 y 150 mg/kg/día) durante la organogénesis, se observó embrioletalidad y disminución del peso corporal materno con la dosis más alta probada. El AUC plasmático para MMF a la dosis sin efecto es aproximadamente 5 veces mayor que en humanos a la DHR.
La administración oral de DMF (25, 100 y 250 mg/kg/día) a ratas durante la organogénesis y la lactancia dio lugar a un aumento de la letalidad, reducciones persistentes en el peso corporal, retraso de la maduración sexual (crías macho y hembra) y reducción del peso testicular a la dosis más alta probada. Se observó deterioro neuroconductual en todas las dosis. No se identificó una dosis sin efecto para la toxicidad del desarrollo. La dosis más baja probada se asoció con un AUC plasmático para MMF inferior al de los humanos a la DHR.
8.2 Lactancia
Resumen de riesgos
No hay datos sobre la presencia de DMF o MMF en la leche humana. Se desconocen los efectos sobre el lactante y la producción de leche.
Se deben considerar los beneficios del desarrollo y la salud de la lactancia materna junto con la necesidad clínica de TECFIDERA de la madre y cualquier posible efecto adverso sobre el lactante amamantado por el medicamento o por la condición materna subyacente.
8.5 Uso geriátrico
Los estudios clínicos de TECFIDERA no incluyeron un número suficiente de pacientes de 65 años o más para determinar si responden de manera diferente a los pacientes más jóvenes.
10 SOBREDOSIS
Se han reportado casos de sobredosis con TECFIDERA. Los síntomas descritos en estos casos fueron consistentes con el perfil de eventos adversos conocido de TECFIDERA.
No se conocen intervenciones terapéuticas para mejorar la eliminación de TECFIDERA ni existe un antídoto conocido. En caso de sobredosis, inicie el tratamiento de apoyo sintomático según esté clínicamente indicado.
11 DESCRIPCIÓN
TECFIDERA contiene dimetilfumarato que también se conoce por su nombre químico, dimetil (E) butendioato, (C6H8O4). Tiene la siguiente estructura:
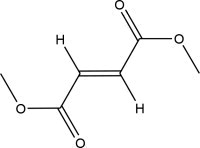
El dimetilfumarato es un polvo blanco a blanco amarillento que es altamente soluble en agua con una masa molecular de 144.13.
TECFIDERA se proporciona en cápsulas de gelatina dura de liberación retardada para administración oral, que contienen 120 mg o 240 mg de dimetilfumarato y constan de los siguientes ingredientes inactivos: celulosa microcristalina, celulosa microcristalina silicificada, croscarmelosa sódica, talco, sílice coloidal dióxido de silicio, estearato de magnesio, citrato de trietilo, copolímero de ácido metacrílico – Tipo A, dispersión de copolímero de ácido metacrílico, simeticona (emulsión al 30 %), laurilsulfato sódico y polisorbato 80. La cápsula, impresa con tinta negra, contiene los siguientes ingredientes inactivos: gelatina, dióxido de titanio, FD&C azul 1; azul brillante FCF, óxido de hierro amarillo y óxido de hierro negro.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Se desconoce el mecanismo por el cual el fumarato de dimetilo (DMF) ejerce su efecto terapéutico en la esclerosis múltiple. Se ha demostrado que el DMF y su metabolito, el fumarato de monometilo (MMF), activan la vía del factor nuclear (eritroide derivado 2) similar a 2 (Nrf2) in vitro e in vivo en animales y humanos. La vía Nrf2 participa en la respuesta celular al estrés oxidativo. Se ha identificado que el MMF es un agonista del receptor de ácido nicotínico in vitro.
12.2 Farmacodinamia
Potencial para prolongar el intervalo QT
En un estudio exhaustivo de QT controlado con placebo realizado en sujetos sanos, no hubo evidencia de que el fumarato de dimetilo causara una prolongación del intervalo QT de importancia clínica (es decir, el límite superior del intervalo de confianza del 90% para el QTc corregido por la línea de base ajustado por placebo fue inferior a 10 ms).
12.3 Farmacocinética
Después de la administración oral de TECFIDERA, el fumarato de dimetilo se somete a una rápida hidrólisis presistemica por las esterasas y se convierte en su metabolito activo, el fumarato de monometilo (MMF). El fumarato de dimetilo no es cuantificable en plasma después de la administración oral de TECFIDERA. Por lo tanto, todos los análisis farmacocinéticos relacionados con TECFIDERA se realizaron con concentraciones plasmáticas de MMF. Los datos farmacocinéticos se obtuvieron en sujetos con esclerosis múltiple y voluntarios sanos.
Absorción
La Tmax mediana de MMF es de 2-2,5 horas. La concentración plasmática máxima (Cmax) y la exposición general (AUC) aumentaron aproximadamente de forma proporcional a la dosis en el rango de dosis estudiado (120 mg a 360 mg). Después de la administración de TECFIDERA 240 mg dos veces al día con alimentos, la Cmax media de MMF fue de 1,87 mg/L y el AUC fue de 8,21 mg.hr/L en pacientes con EM.
Una comida rica en grasas y calorías no afectó el AUC de MMF, pero disminuyó su Cmax en un 40%. La Tmax se retrasó de 2,0 horas a 5,5 horas. En este estudio, la incidencia de rubor se redujo aproximadamente un 25% en estado de alimentación.
Distribución
El volumen de distribución aparente de MMF varía entre 53 y 73 L en sujetos sanos. La unión a proteínas plasmáticas humanas de MMF es del 27-45% e independiente de la concentración.
Metabolismo
En humanos, el fumarato de dimetilo se metaboliza ampliamente por las esterasas, que son ubicuas en el tracto gastrointestinal, la sangre y los tejidos, antes de que llegue a la circulación sistémica. El metabolismo adicional de MMF se produce a través del ciclo del ácido tricarboxílico (TCA), sin la participación del sistema del citocromo P450 (CYP). MMF, ácido fumárico y cítrico, y glucosa son los principales metabolitos en plasma.
Eliminación
La exhalación de CO2 es la principal vía de eliminación, representando aproximadamente el 60% de la dosis de TECFIDERA. La eliminación renal y fecal son vías menores de eliminación, representando el 16% y el 1% de la dosis, respectivamente. Se encontraron cantidades traza de MMF sin cambios en la orina.
La vida media terminal de MMF es de aproximadamente 1 hora y no hay MMF circulante presente a las 24 horas en la mayoría de los individuos. No se produce acumulación de MMF con dosis múltiples de TECFIDERA.
Poblaciones específicas
El peso corporal, el sexo y la edad no requieren ajuste de la dosis.
No se han realizado estudios en sujetos con insuficiencia hepática o renal. Sin embargo, no se esperaría que ninguna de las dos afecciones afectara la exposición a MMF y, por lo tanto, no es necesario ningún ajuste de la dosis.
Estudios de interacción medicamentosa
No se identificaron posibles interacciones medicamentosas con el fumarato de dimetilo o MMF en estudios de inhibición e inducción de CYP in vitro, o en estudios de glucoproteína P. Las dosis únicas de interferón beta-1a o acetato de glatiramer no alteraron la farmacocinética de MMF. La aspirina, cuando se administra aproximadamente 30 minutos antes de TECFIDERA, no alteró la farmacocinética de MMF.
Anticonceptivos orales
La coadministración de fumarato de dimetilo con un anticonceptivo oral combinado (norelgestromina y etinilestradiol) no provocó ningún efecto relevante en la exposición a los anticonceptivos orales. No se han realizado estudios de interacción con anticonceptivos orales que contengan otros progestágenos.
Vacunas
Un estudio aleatorizado, abierto, examinó el uso concomitante de TECFIDERA y varias vacunas no vivas en adultos de 27 a 55 años de edad con formas recurrentes de EM (38 sujetos que se sometieron a tratamiento con TECFIDERA en el momento de la vacunación y 33 sujetos que se sometieron a tratamiento con interferón no peguilado en el momento de la vacunación). La exposición concomitante a TECFIDERA no atenuó las respuestas de anticuerpos a la vacuna que contiene toxoide tetánico, polisacárido neumocócico y vacunas meningocócicas en relación con las respuestas de anticuerpos en pacientes tratados con interferón. Se desconoce el impacto de estos hallazgos en la eficacia de la vacuna en esta población de pacientes. No se ha evaluado la seguridad y eficacia de las vacunas vivas o atenuadas vivas administradas de forma concomitante con TECFIDERA.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
Carcinogénesis
Se realizaron estudios de carcinogenicidad de fumarato de dimetilo (DMF) en ratones y ratas. En ratones, la administración oral de DMF (25, 75, 200 y 400 mg/kg/día) durante un máximo de dos años dio como resultado un aumento en los tumores del estómago no glandular (preestómago) y los riñones: carcinomas de células escamosas y papilomas del preestómago en machos y hembras a 200 y 400 mg/kg/día; leiomiosarcomas del preestómago a 400 mg/kg/día en machos y hembras; adenomas y carcinomas tubulares renales a 200 y 400 mg/kg/día en machos; y adenomas del túbulo renal a 400 mg/kg/día en hembras. La exposición plasmática a MMF (AUC) en la dosis más alta no asociada con tumores en ratones (75 mg/kg/día) fue similar a la de los humanos a la dosis humana recomendada (RHD) de 480 mg/día.
En ratas, la administración oral de DMF (25, 50, 100 y 150 mg/kg/día) durante un máximo de dos años dio como resultado aumentos en los carcinomas de células escamosas y papilomas del preestómago a todas las dosis probadas en machos y hembras, y en adenomas de células intersticiales (Leydig) testiculares a 100 y 150 mg/kg/día. El AUC plasmático de MMF a la dosis más baja probada fue menor que la de los humanos a la RHD.
Mutagénesis
El fumarato de dimetilo (DMF) y el fumarato de monometilo (MMF) no fueron mutagénicos en la prueba de mutación inversa bacteriana in vitro (Ames). DMF y MMF fueron clastogénicos en la prueba de aberración cromosómica in vitro en linfocitos de sangre periférica humana en ausencia de activación metabólica. DMF no fue clastogénico en la prueba de micronúcleos in vivo en la rata.
Deterioro de la Fertilidad
En ratas macho, la administración oral de DMF (75, 250 y 375 mg/kg/día) antes y durante todo el período de apareamiento no tuvo ningún efecto sobre la fertilidad; sin embargo, se observaron aumentos en el esperma no móvil a las dosis media y alta. La dosis sin efecto para los efectos adversos sobre el esperma es similar a la dosis humana recomendada (RHD) de 480 mg/día en base al área de superficie corporal (mg/m2).
En ratas hembra, la administración oral de DMF (20, 100 y 250 mg/kg/día) antes y durante el apareamiento y continuando hasta el día 7 de gestación causó una interrupción del ciclo estral y aumentos en la embrioletalidad a la dosis más alta probada. La dosis más alta no asociada con efectos adversos (100 mg/kg/día) es el doble de la RHD en base a mg/m2.
Se observó toxicidad testicular (degeneración del epitelio germinal, atrofia, hipospermia y/o hiperplasia) a dosis clínicamente relevantes en ratones, ratas y perros en estudios de toxicidad oral subcrónica y crónica de DMF, y en un estudio de toxicidad oral crónica que evaluó una combinación de cuatro ésteres de ácido fumárico (incluido DMF) en ratas.
13.2 Toxicología y/o Farmacología Animal
Se observó toxicidad renal después de la administración oral repetida de fumarato de dimetilo (DMF) en ratones, ratas, perros y monos. Se observó regeneración del epitelio del túbulo renal, sugestiva de lesión del epitelio del túbulo, en todas las especies. Se observó hiperplasia tubular renal en ratas con dosificación de hasta dos años. Se observó atrofia cortical y fibrosis intersticial en perros y monos a dosis superiores a 5 mg/kg/día. En monos, la dosis más alta probada (75 mg/kg/día) se asoció con necrosis de células individuales y fibrosis intersticial multifocal y difusa, lo que indica una pérdida irreversible de tejido renal y función. En perros y monos, la dosis de 5 mg/kg/día se asoció con exposiciones plasmáticas a MMF menores o similares a las de los humanos a la dosis humana recomendada (RHD).
Se observó un aumento relacionado con la dosis en la incidencia y gravedad de la degeneración retiniana en ratones después de la administración oral de DMF durante un máximo de dos años a dosis superiores a 75 mg/kg/día, una dosis asociada con una exposición plasmática a MMF (AUC) similar a la de los humanos a la RHD.
14 ESTUDIOS CLÍNICOS
La eficacia y seguridad de TECFIDERA se demostraron en dos estudios (Estudios 1 y 2) que evaluaron TECFIDERA tomado dos o tres veces al día en pacientes con esclerosis múltiple remitente-recurrente (EMRR). La dosis inicial de TECFIDERA fue de 120 mg dos o tres veces al día durante los primeros 7 días, seguida de un aumento a 240 mg dos o tres veces al día. Ambos estudios incluyeron pacientes que habían experimentado al menos 1 recaída durante el año anterior al ensayo o que tenían una resonancia magnética (RM) del cerebro que demostraba al menos una lesión que realzaba con gadolinio (Gd+) dentro de las 6 semanas de la aleatorización. La Escala Expandida del Estado de Discapacidad (EDSS) también se evaluó y los pacientes podrían tener puntuaciones que van de 0 a 5. Se realizaron evaluaciones neurológicas al inicio, cada 3 meses y en el momento de una recaída sospechosa. Las evaluaciones de RM se realizaron al inicio, al mes 6 y al año 1 y 2 en un subconjunto de pacientes (44% en el Estudio 1 y 48% en el Estudio 2).
Estudio 1: Ensayo controlado con placebo en EMRR
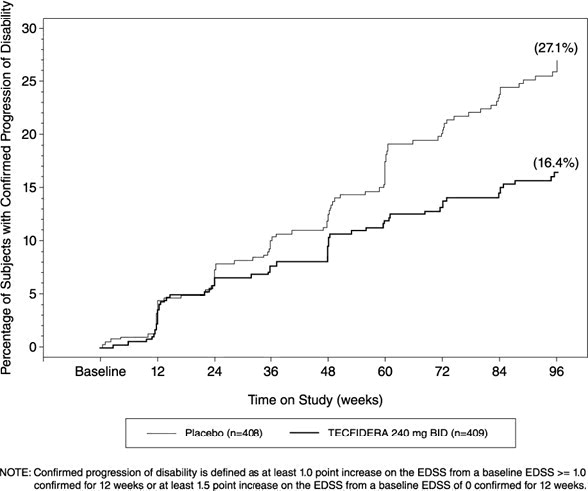
El Estudio 1 fue un estudio de 2 años, aleatorizado, doble ciego, controlado con placebo en 1234 pacientes con EMRR. El criterio de valoración principal fue la proporción de pacientes que recayeron a los 2 años. Los criterios de valoración adicionales a los 2 años incluyeron el número de lesiones nuevas o de reciente aumento de tamaño hiperintensas en T2, el número de lesiones nuevas hipointensas en T1, el número de lesiones Gd+, la tasa de recaída anualizada (TRA) y el tiempo hasta la progresión de la discapacidad confirmada. La progresión de la discapacidad confirmada se definió como un aumento de al menos 1 punto desde la EDSS basal (aumento de 1,5 puntos para los pacientes con EDSS basal de 0) sostenido durante 12 semanas.
Los pacientes fueron aleatorizados para recibir TECFIDERA 240 mg dos veces al día (n=410), TECFIDERA 240 mg tres veces al día (n=416) o placebo (n=408) durante un máximo de 2 años. La edad media fue de 39 años, el tiempo medio desde el diagnóstico fue de 4 años y la puntuación media de EDSS al inicio fue de 2. El tiempo medio con el fármaco del estudio para todos los brazos de tratamiento fue de 96 semanas. Los porcentajes de pacientes que completaron 96 semanas con el fármaco del estudio por grupo de tratamiento fueron del 69% para los pacientes asignados a TECFIDERA 240 mg dos veces al día, del 69% para los pacientes asignados a TECFIDERA 240 mg tres veces al día y del 65% para los pacientes asignados a los grupos placebo.
TECFIDERA tuvo un efecto estadísticamente significativo en todos los criterios de valoración descritos anteriormente y la dosis de 240 mg tres veces al día no mostró ningún beneficio adicional sobre la dosis de TECFIDERA 240 mg dos veces al día. Los resultados de este estudio (240 mg dos veces al día frente a placebo) se muestran en Tabla 2 y Figura 1.
| TECFIDERA 240 mg BID |
Placebo |
P-value |
|
|---|---|---|---|
| Criterios de valoración clínicos | N=410 | N=408 | |
| Proporción de recaídas (criterio de valoración principal) Reducción del riesgo relativo |
27% 49% |
46% | <0.0001 |
| Tasa de recaída anualizada | 0.172 | 0.364 | <0.0001 |
| Reducción relativa | 53% | ||
| Proporción con progresión de la discapacidad | 16% | 27% | 0.0050 |
| Reducción del riesgo relativo | 38% | ||
| Criterios de valoración de RM | N=152 | N=165 | |
| Número medio de lesiones nuevas o de reciente aumento de tamaño | 2.6 | 17 | <0.0001 |
| lesiones en T2 durante 2 años | |||
| Porcentaje de sujetos sin lesiones nuevas o de reciente aumento de tamaño en T2 |
45% | 27% | |
| Número de lesiones Gd+ a los 2 años | 0.1 (0) | 1.8 (0) | |
| Media (mediana) | |||
| Porcentaje de sujetos con | |||
| 0 lesiones | 93% | 62% | |
| 1 lesión | 5% | 10% | |
| 2 lesiones | <1% | 8% | |
| 3 a 4 lesiones | 0 | 9% | |
| 5 o más lesiones | <1% | 11% | |
| Reducción de las probabilidades relativas | 90% | <0.0001 | |
| (porcentaje) | |||
| Número medio de nuevas lesiones T1 hipointensas | 1.5 | 5.6 | <0.0001 |
| durante 2 años |
Figura 1: Tiempo hasta la progresión confirmada de la discapacidad a las 12 semanas (Estudio 1)
Estudio 2: Ensayo controlado con placebo en RRMS
El estudio 2 fue un estudio multicéntrico, aleatorizado, doble ciego, controlado con placebo de 2 años que también incluyó un brazo de comparación de etiqueta abierta en pacientes con RRMS. El criterio de valoración principal fue la tasa de recaídas anualizada a los 2 años. Los criterios de valoración adicionales a los 2 años incluyeron el número de lesiones T2 hiperintensas nuevas o de reciente aumento de tamaño, el número de lesiones T1 hipointensas, el número de lesiones Gd+, la proporción de pacientes que recayeron y el tiempo hasta la progresión confirmada de la discapacidad según se definió en el estudio 1.
Los pacientes fueron aleatorizados para recibir TECFIDERA 240 mg dos veces al día (n=359), TECFIDERA 240 mg tres veces al día (n=345), un comparador de etiqueta abierta (n=350) o placebo (n=363) durante un máximo de 2 años. La edad media fue de 37 años, el tiempo medio desde el diagnóstico fue de 3 años y la puntuación EDSS media en el momento basal fue de 2,5. El tiempo medio en el fármaco del estudio para todos los brazos de tratamiento fue de 96 semanas. Los porcentajes de pacientes que completaron 96 semanas en el fármaco del estudio por grupo de tratamiento fueron del 70% para los pacientes asignados a TECFIDERA 240 mg dos veces al día, del 72% para los pacientes asignados a TECFIDERA 240 mg tres veces al día y del 64% para los pacientes asignados a los grupos placebo.
TECFIDERA tuvo un efecto estadísticamente significativo en los criterios de valoración de recaídas y RM descritos anteriormente. No hubo un efecto estadísticamente significativo en la progresión de la discapacidad. La dosis de TECFIDERA 240 mg tres veces al día no produjo ningún beneficio adicional sobre la dosis de TECFIDERA 240 mg dos veces al día. Los resultados de este estudio (240 mg dos veces al día frente a placebo) se muestran en Tabla 3.
| TECFIDERA 240 mg BID |
Placebo |
Valor de p |
|
|---|---|---|---|
| Criterios de valoración clínicos | N=359 | N=363 | |
| Tasa de recaídas anualizada | 0,224 | 0,401 | <0,0001 |
| Reducción relativa | 44% | ||
| Proporción de pacientes que recayeron | 29% | 41% | 0,0020 |
| Reducción del riesgo relativo | 34% | ||
| Proporción de pacientes con progresión de la discapacidad | 13% | 17% | 0,25 |
| Reducción del riesgo relativo | 21% | ||
| Criterios de valoración de RM | N=147 | N=144 | |
| Número medio de lesiones T2 nuevas o de reciente aumento de tamaño | 5,1 | 17,4 | <0,0001 |
| durante 2 años | |||
| Porcentaje de sujetos sin lesiones nuevas o | 27% | 12% | |
| de reciente aumento de tamaño | |||
| Número de lesiones Gd+ a los 2 años | |||
| Media (mediana) | 0.5 (0.0) | 2.0 (0.0) | |
| Porcentaje de sujetos con | |||
| 0 lesiones | 80% | 61% | |
| 1 lesión | 11% | 17% | |
| 2 lesiones | 3% | 6% | |
| 3 a 4 lesiones | 3% | 2% | |
| 5 o más lesiones | 3% | 14% | |
| Reducción de la probabilidad relativa | 74% | <0.0001 | |
| (porcentaje) | |||
| Número medio de nuevas lesiones T1 hipointensas | 3.0 | 7.0 | <0.0001 |
| durante 2 años |
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
TECFIDERA está disponible como cápsulas de liberación retardada de gelatina dura en dos concentraciones que contienen 120 mg o 240 mg de fumarato de dimetilo. Las cápsulas verdes y blancas de 120 mg están impresas con “BG-12 120 mg” en tinta negra. Las cápsulas verdes de 240 mg están impresas con “BG-12 240 mg” en tinta negra. TECFIDERA está disponible de la siguiente manera:
Paquete de inicio de 30 días, (NDC 64406-007-03):
- Frasco de 7 días de cápsulas de 120 mg, cantidad 14
- Frasco de 23 días de cápsulas de 240 mg, cantidad 46
Cápsulas de 120 mg:
- Frasco de 7 días de 14 cápsulas (NDC 64406-005-01)
Cápsulas de 240 mg:
- Frasco de 30 días de 60 cápsulas (NDC 64406-006-02)
Almacenar a 15°C a 30°C (59 a 86°F). Proteger las cápsulas de la luz. Almacenar en el envase original.
17 INFORMACIÓN PARA EL PACIENTE
Aconseje al paciente que lea el etiquetado del paciente aprobado por la FDA (Información para el paciente).
Dosificación
Informe a los pacientes que se les proporcionarán dos concentraciones de TECFIDERA al comenzar el tratamiento: cápsulas de 120 mg para la dosis inicial de 7 días y cápsulas de 240 mg para la dosis de mantenimiento, ambas se tomarán dos veces al día. Informe a los pacientes que deben tragar las cápsulas de TECFIDERA enteras e intactas. Informe a los pacientes que no deben triturar, masticar o espolvorear el contenido de las cápsulas sobre los alimentos. Informe a los pacientes que TECFIDERA se puede tomar con o sin alimentos [ver Dosificación y administración (2.1)].
Anafilaxia y Angioedema
Aconseje a los pacientes que suspendan TECFIDERA y busquen atención médica si desarrollan signos y síntomas de anafilaxia o angioedema [ver Advertencias y precauciones (5.1)].
Leucoencefalopatía Multifocal Progresiva
Informe a los pacientes que se ha producido leucoencefalopatía multifocal progresiva (PML) en pacientes que recibieron TECFIDERA. Informe al paciente que la PML se caracteriza por una progresión de déficits y generalmente conduce a la muerte o discapacidad grave en semanas o meses. Indique al paciente la importancia de ponerse en contacto con su médico si desarrolla algún síntoma sugestivo de PML. Informe al paciente que los síntomas típicos asociados con la PML son diversos, progresan durante días o semanas e incluyen debilidad progresiva en un lado del cuerpo o torpeza de las extremidades, alteración de la visión y cambios en el pensamiento, la memoria y la orientación que conducen a confusión y cambios de personalidad [ver Advertencias y precauciones (5.2)].
Herpes Zoster y otras infecciones oportunistas graves
Informe a los pacientes que se ha producido herpes zoster y otras infecciones oportunistas graves en pacientes que recibieron TECFIDERA. Indique al paciente la importancia de ponerse en contacto con su médico si desarrolla algún signo o síntoma asociado con herpes zoster u otras infecciones oportunistas graves [ver Advertencias y precauciones (5.3)].
Recuentos de linfocitos
Informe a los pacientes que TECFIDERA puede disminuir los recuentos de linfocitos. Se debe obtener un análisis de sangre antes de comenzar la terapia. También se recomiendan análisis de sangre después de 6 meses de tratamiento, cada 6 a 12 meses a partir de entonces y según esté clínicamente indicado [ver Advertencias y precauciones (5.3), Reacciones adversas (6.1)].
Lesión hepática
Informe a los pacientes que TECFIDERA puede causar lesión hepática. Indique a los pacientes tratados con TECFIDERA que informen de inmediato cualquier síntoma que pueda indicar lesión hepática, incluida la fatiga, la anorexia, el dolor en el cuadrante superior derecho del abdomen, la orina oscura o la ictericia. Se debe obtener un análisis de sangre antes de que los pacientes comiencen la terapia y durante el tratamiento, según esté clínicamente indicado [ver Advertencias y precauciones (5.4)].
Rubor
Informe a los pacientes que el rubor es una de las reacciones más comunes, especialmente al inicio de la terapia, y puede disminuir con el tiempo. Aconseje a los pacientes que se pongan en contacto con su proveedor de atención médica si experimentan rubor persistente y/o grave. Aconseje a los pacientes que experimentan rubor que tomar TECFIDERA con alimentos o tomar una aspirina no recubierta entérica antes de tomar TECFIDERA puede ayudar [ver Reacciones adversas (6.1)].
Eventos gastrointestinales (GI)
Informe a los pacientes que los eventos GI (dolor abdominal, diarrea y náuseas) son algunas de las reacciones adversas más comunes, especialmente al inicio de la terapia, y pueden disminuir con el tiempo. Algunos pacientes pueden experimentar eventos GI más graves. Aconseje a los pacientes que se pongan en contacto con su proveedor de atención médica de inmediato y suspendan TECFIDERA si experimentan sangrado gastrointestinal (por ejemplo, sangrado rectal, diarrea con sangre, hematemesis) u otros eventos adversos gastrointestinales graves (por ejemplo, dolor abdominal intenso, vómitos y/o diarrea intensos) [ver Advertencias y precauciones (5.7)].
PPI-41347-18
Fabricado para:
Biogen Inc.
Cambridge, MA 02142
TECFIDERA es una marca registrada de Biogen.
© Biogen 2013 – 2024
INSERTO PARA EL PACIENTE
|
Esta información para pacientes ha sido aprobada por la Administración de Alimentos y Medicamentos de los Estados Unidos. Revisado: 03/2024 |
|
PPI-41347-3 |
| Información para pacientes TECFIDERA® (tek” fi de’ rah) (cápsulas de liberación retardada de dimetil fumarato) |
¿Qué es TECFIDERA?
|
¿Quién no debe tomar TECFIDERA?
|
Antes de tomar y mientras tome TECFIDERA, informe a su médico si tiene o ha tenido:
|
Informe a su médico si usted es:
|
¿Cómo debo tomar TECFIDERA?
|
| ¿Cuáles son los posibles efectos secundarios de TECFIDERA? TECFIDERA puede causar efectos secundarios graves, incluyendo:
|
Los efectos secundarios más comunes de TECFIDERA incluyen:
|
| Estos no son todos los posibles efectos secundarios de TECFIDERA. Llame a su médico para obtener consejo médico sobre los efectos secundarios. Puede reportar los efectos secundarios a la FDA al 1 – 800 – FDA – 1088. Para más información vaya a dailymed.nlm.nih.gov. |
Información general sobre el uso seguro y eficaz de TECFIDERA
|
| ¿Cuáles son los ingredientes de TECFIDERA? Ingrediente activo: dimetil fumarato Ingredientes inactivos: celulosa microcristalina, celulosa microcristalina silicificada, sodio croscarmelosa, talco, dióxido de silicio coloidal, estearato de magnesio, citrato de trietilo, copolímero de ácido metacrílico – Tipo A, dispersión de copolímero de ácido metacrílico, simeticona (emulsión al 30%), laurilsulfato sódico y polisorbato 80. Cápsula: gelatina, dióxido de titanio, azul FD&C 1; azul brillante FCF, óxido de hierro amarillo y óxido de hierro negro.Fabricado para: Biogen Inc., Cambridge, MA 02142, www.TECFIDERA.com o llame al 1-800-456-2255 |
PANEL DE VISUALIZACIÓN PRINCIPAL
Panel de exhibición principal – Cápsulas de 240 mg: Etiqueta de la caja
NDC 64406-006-02
60 cápsulas
Tecfidera®
(dimetil fumarato)
cápsulas de liberación retardada
240 mg
Dispensar en el envase original.
Tragar la cápsula entera.
Solo con receta médica

PANEL DE VISUALIZACIÓN PRINCIPAL
Principal Display Panel – 120 mg Capsules: Box Label
NDC 64406-005-01
14 capsules
Tecfidera®
(dimethyl fumarate)
delayed-release capsules
120 mg
Dispense in original Package.
Swallow capsule whole.
Rx only

Panel de visualización principal – Cápsulas de 120 mg: Etiqueta de caja sin cargo
NDC 64406-005-08
14 cápsulas
Tecfidera®
(dimetil fumarato)
cápsulas de liberación retardada
120 mg
Dispensar en el envase original.
Tragar la cápsula entera.
Solo con receta médica

Panel de Presentación Principal – Cápsulas de 120 mg: Etiqueta de la Caja de Muestra
NDC 64406-005-10
14 cápsulas
Tecfidera®
(dimetil fumarato)
cápsulas de liberación retardada
120 mg
Dispensar en el envase original.
Tragar la cápsula entera.
Solo con receta médica

PANEL DE VISUALIZACIÓN PRINCIPAL
Panel principal de presentación – Paquete inicial: Etiqueta de la caja
NDC 64406-007-03
Paquete inicial de 30 días
Tecfidera®
(dimetil fumarato)
cápsulas de liberación retardada
Contenido del paquete:
Dosis inicial de 120 mg
Un frasco que contiene 14
cápsulas de 120 mg cada una
Dosis regular de 240 mg
Un frasco que contiene 46
cápsulas de 240 mg cada una
Dispensar en el envase original.
Consulte el panel posterior para obtener información sobre la dosificación y la administración
instrucciones de uso.
