
Fabricante de medicamentos: Amgen Inc (Updated: 2024-01-12)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
Xgeva (denosumab) injection, para uso subcutáneo
Aprobación inicial en EE. UU.: 2010
CAMBIOS RECIENTES IMPORTANTES
| Advertencias y precauciones, Hipocalcemia (5.3) | 02/2020 |
INDICACIONES Y USO
Xgeva es un inhibidor del ligando RANK (RANKL) indicado para:
- Prevención de eventos esqueléticos relacionados en pacientes con mieloma múltiple y en pacientes con metástasis óseas de tumores sólidos. (1.1)
- Tratamiento de adultos y adolescentes esqueléticamente maduros con tumor de células gigantes del hueso que es irresecable o donde la resección quirúrgica es probable que resulte en morbilidad grave. (1.2, 14.3)
- Tratamiento de la hipercalcemia de malignidad refractaria al tratamiento con bisfosfonatos. (1.3)
DOSIFICACIÓN Y ADMINISTRACIÓN
- Xgeva está destinado únicamente para la vía subcutánea y no debe administrarse por vía intravenosa, intramuscular o intradérmica. (2.1)
- Mieloma múltiple y metástasis ósea de tumores sólidos: Administrar 120 mg cada 4 semanas como una inyección subcutánea en la parte superior del brazo, la parte superior del muslo o el abdomen. (2.2)
- Tumor de células gigantes del hueso: Administrar 120 mg cada 4 semanas con dosis adicionales de 120 mg en los días 8 y 15 del primer mes de terapia. Administrar por vía subcutánea en la parte superior del brazo, la parte superior del muslo o el abdomen. (2.3)
- Administrar calcio y vitamina D según sea necesario para tratar o prevenir la hipocalcemia. (2.2, 2.3)
- Hipercalcemia de malignidad: Administrar 120 mg cada 4 semanas con dosis adicionales de 120 mg en los días 8 y 15 del primer mes de terapia. Administrar por vía subcutánea en la parte superior del brazo, la parte superior del muslo o el abdomen. (2.4)
FORMAS DE DOSIFICACIÓN Y FUERZAS
- Inyección: 120 mg/1.7 mL (70 mg/mL) solución en un vial de dosis única (3)
CONTRAINDICACIONES
ADVERTENCIAS Y PRECAUCIONES
- Mismo ingrediente activo: Los pacientes que reciben Xgeva no deben tomar Prolia®. (5.1)
- Pueden ocurrir reacciones de hipersensibilidad, incluida la anafilaxia. Suspenda permanentemente si ocurre una reacción clínicamente significativa. (5.2)
- Hipocalcemia: Xgeva puede causar hipocalcemia sintomática grave, y se han reportado casos fatales. Corrija la hipocalcemia antes de iniciar Xgeva. Controle los niveles de calcio durante la terapia, especialmente en las primeras semanas de inicio de la terapia, y suplemente adecuadamente a todos los pacientes con calcio y vitamina D. (5.3)
- Se ha reportado osteonecrosis de la mandíbula (ONJ) en pacientes que reciben Xgeva. Realice un examen oral antes de comenzar Xgeva. Controle los síntomas. Evite los procedimientos dentales invasivos durante el tratamiento con Xgeva. (5.4)
- Fractura femoral atípica: Evalúe a los pacientes con dolor en el muslo o la ingle para descartar una fractura femoral. (5.5)
- Hipercalcemia después de la interrupción del tratamiento en pacientes con tumor de células gigantes del hueso y en pacientes con esqueletos en crecimiento: Controle a los pacientes para detectar signos y síntomas de hipercalcemia y maneje según sea clínicamente apropiado. (5.6, 8.4)
- Fracturas vertebrales múltiples (MVF) después de la interrupción del tratamiento: Cuando se interrumpe el tratamiento con Xgeva, evalúe el riesgo individual del paciente de fracturas vertebrales. (5.7)
- Toxicidad embrio-fetal: Puede causar daño fetal. Avise a las mujeres en edad fértil del riesgo potencial para el feto y de usar métodos anticonceptivos efectivos. (5.8, 8.1, 8.3)
REACCIONES ADVERSAS
- Metástasis ósea de tumores sólidos: Las reacciones adversas más comunes (≥ 25%) fueron fatiga/astenia, hipofosfatemia y náuseas. (6.1)
- Mieloma múltiple: Las reacciones adversas más comunes (≥ 10%) fueron diarrea, náuseas, anemia, dolor de espalda, trombocitopenia, edema periférico, hipocalcemia, infección del tracto respiratorio superior, erupción cutánea y dolor de cabeza. (6.1)
- Tumor de células gigantes del hueso: Las reacciones adversas más comunes (≥ 10%) fueron artralgia, dolor de cabeza, náuseas, dolor de espalda, fatiga y dolor en las extremidades. (6.1)
- Hipercalcemia de malignidad: Las reacciones adversas más comunes (> 20%) fueron náuseas, disnea, disminución del apetito, dolor de cabeza, edema periférico, vómitos, anemia, estreñimiento y diarrea. (6.1)
Para reportar REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Amgen Inc. al 1-800-77-AMGEN (1-800-772-6436) o la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
USO EN POBLACIONES ESPECÍFICAS
- Pacientes pediátricos: Recomendado solo para el tratamiento de adolescentes esqueléticamente maduros con tumor de células gigantes del hueso. (8.4)
- Insuficiencia renal: Los pacientes con aclaramiento de creatinina inferior a 30 mL/min o que reciben diálisis tienen riesgo de hipocalcemia. Suplemente adecuadamente con calcio y vitamina D. (8.6)
Ver 17 para INFORMACIÓN DE ASESORAMIENTO AL PACIENTE.
Revisado: 6/2020
Tabla de Contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
1.1 Mieloma Múltiple y Metástasis Ósea de Tumores Sólidos
1.2 Tumor de Células Gigantes del Hueso
1.3 Hipercalcemia de la Malignidad
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Instrucciones Importantes de Administración
2.2 Mieloma Múltiple y Metástasis Ósea de Tumores Sólidos
2.3 Tumor de Células Gigantes del Hueso
2.4 Hipercalcemia de la Malignidad
2.5 Preparación y Administración
3 FORMAS Y FUERZAS DE DOSIFICACIÓN
4 CONTRAINDICACIONES
4.1 Hipocalcemia
4.2 Hipersensibilidad
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Productos Farmacéuticos con el Mismo Ingrediente Activo
5.2 Hipersensibilidad
5.3 Hipocalcemia
5.4 Osteonecrosis de la Mandíbula (ONJ)
5.5 Fractura Femoral Atípica Subtrocantérica y Diafisaria
5.6 Hipercalcemia Después de la Suspensión del Tratamiento en Pacientes con Tumor de Células Gigantes del Hueso y en Pacientes con Esqueletos en Crecimiento
5.7 Fracturas Vertebrales Múltiples (MVF) Después de la Suspensión del Tratamiento
5.8 Toxicidad Embriofetal
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos
6.2 Experiencia Postcomercialización
6.3 Inmunogenicidad
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y Hombres en Potencial Reproductivo
8.4 Uso Pediátrico
8.5 Uso Geriátrico
8.6 Insuficiencia Renal
10 SOBREDOSIS
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
13.2 Toxicología y/o Farmacología Animal
14 ENSAYOS CLÍNICOS
14.1 Metástasis Ósea de Tumores Sólidos
14.2 Mieloma Múltiple
14.3 Tumor de Células Gigantes del Hueso
14.4 Hipercalcemia de la Malignidad
16 CÓMO SE SUMINISTRA/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no están enumeradas.
1 INDICACIONES Y USO
1.1 Mieloma Múltiple y Metástasis Ósea de Tumores Sólidos
Xgeva está indicado para la prevención de eventos esqueléticos relacionados en pacientes con mieloma múltiple y en pacientes con metástasis óseas de tumores sólidos.
1.2 Tumor de Células Gigantes del Hueso
Xgeva está indicado para el tratamiento de adultos y adolescentes esqueléticamente maduros con tumor de células gigantes del hueso que es irresecable o donde la resección quirúrgica es probable que resulte en morbilidad grave [ver Ensayos Clínicos (14.2)].
1.3 Hipercalcemia de la Malignidad
Xgeva está indicado para el tratamiento de la hipercalcemia de la malignidad refractaria a la terapia con bisfosfonatos.
2 DOSIS Y ADMINISTRACIÓN
2.1 Instrucciones Importantes de Administración
Xgeva está destinado únicamente para la vía subcutánea y no debe administrarse por vía intravenosa, intramuscular o intradérmica.
2.2 Mieloma Múltiple y Metástasis Ósea de Tumores Sólidos
La dosis recomendada de Xgeva es de 120 mg administrada como inyección subcutánea cada 4 semanas en la parte superior del brazo, la parte superior del muslo o el abdomen.
Administrar calcio y vitamina D según sea necesario para tratar o prevenir la hipocalcemia [ver Advertencias y precauciones (5.3)].
2.3 Tumor de Células Gigantes del Hueso
La dosis recomendada de Xgeva es de 120 mg administrada cada 4 semanas con dosis adicionales de 120 mg en los Días 8 y 15 del primer mes de terapia. Administrar por vía subcutánea en la parte superior del brazo, la parte superior del muslo o el abdomen.
Administrar calcio y vitamina D según sea necesario para tratar o prevenir la hipocalcemia [ver Advertencias y precauciones (5.3)].
2.4 Hipercalcemia de la Malignidad
La dosis recomendada de Xgeva es de 120 mg administrada cada 4 semanas con dosis adicionales de 120 mg en los Días 8 y 15 del primer mes de terapia. Administrar por vía subcutánea en la parte superior del brazo, la parte superior del muslo o el abdomen.
2.5 Preparación y Administración
Inspeccione visualmente Xgeva para detectar partículas y decoloración antes de la administración. Xgeva es una solución transparente, incolora a amarillo pálido que puede contener trazas de partículas proteicas traslúcidas a blancas. No lo use si la solución está decolorada o turbia o si la solución contiene muchas partículas o materia particulada extraña.
Antes de la administración, Xgeva puede retirarse del refrigerador y llevarse a temperatura ambiente (hasta 25°C/77°F) colocándolo en el envase original. Esto generalmente toma de 15 a 30 minutos. No caliente Xgeva de ninguna otra manera [ver Cómo se suministra/Almacenamiento y manipulación (16)].
Use una aguja de calibre 27 para extraer e inyectar todo el contenido del vial. No vuelva a entrar en el vial. Deseche el vial después de una sola dosis o entrada.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Inyección: 120 mg/1.7 mL (70 mg/mL) solución en un vial de dosis única.
4 CONTRAINDICACIONES
4.1 Hipocalcemia
La hipocalcemia preexistente debe corregirse antes de iniciar el tratamiento con Xgeva [ver Advertencias y precauciones (5.3)].
4.2 Hipersensibilidad
Xgeva está contraindicado en pacientes con hipersensibilidad clínicamente significativa conocida a Xgeva [ver Advertencias y precauciones (5.2) y Reacciones adversas (6.2)].
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Productos farmacéuticos con el mismo ingrediente activo
Xgeva incluye el mismo ingrediente activo (denosumab) que se encuentra en Prolia. Los pacientes que reciben Xgeva no deben tomar Prolia.
5.2 Hipersensibilidad
Se ha informado de hipersensibilidad clínicamente significativa, incluida la anafilaxia, con el uso de Xgeva. Las reacciones pueden incluir hipotensión, disnea, edema de las vías respiratorias superiores, hinchazón de los labios, erupción cutánea, prurito y urticaria. Si se produce una reacción alérgica anafiláctica u otra clínicamente significativa, inicie la terapia adecuada y suspenda el tratamiento con Xgeva de forma permanente [see Contraindications (4.2) y Adverse Reactions (6.2)].
5.3 Hipocalcemia
Xgeva puede causar hipocalcemia sintomática grave, y se han notificado casos mortales. Corrija la hipocalcemia preexistente antes del tratamiento con Xgeva. Controle los niveles de calcio durante todo el tratamiento con Xgeva, especialmente en las primeras semanas de inicio del tratamiento, y administre calcio, magnesio y vitamina D según sea necesario. El uso concomitante de calcimiméticos y otros medicamentos que pueden reducir los niveles de calcio puede empeorar el riesgo de hipocalcemia y se debe controlar estrechamente el calcio sérico. Avise a los pacientes que se pongan en contacto con un profesional sanitario si presentan síntomas de hipocalcemia [see Contraindications (4.1), Adverse Reactions (6.1, 6.2), and Patient Counseling Information (17)].
Se ha observado un mayor riesgo de hipocalcemia en los ensayos clínicos de pacientes con disfunción renal creciente, más comúnmente con disfunción grave (aclaramiento de creatinina inferior a 30 mL/min y/o en diálisis), y con una suplementación de calcio inadecuada/nula. Controle los niveles de calcio y la ingesta de calcio y vitamina D [see Adverse Reactions (6.1), Use in Specific Populations (8.6), and Clinical Pharmacology (12.3)].
5.4 Osteonecrosis de la mandíbula (ONJ)
Se ha notificado osteonecrosis de la mandíbula (ONJ) en pacientes que reciben Xgeva, que se manifiesta como dolor en la mandíbula, osteomielitis, osteítis, erosión ósea, infección dental o periodontal, dolor de muelas, ulceración gingival o erosión gingival. El dolor persistente o la curación lenta de la boca o la mandíbula después de una cirugía dental también pueden ser manifestaciones de ONJ. En los ensayos clínicos en pacientes con cáncer, la incidencia de ONJ fue mayor con una duración de exposición más larga [see Adverse Reactions (6.1)]. El setenta y nueve por ciento de los pacientes con ONJ tenían antecedentes de extracción dental, mala higiene oral o uso de un aparato dental como factor predisponente. Otros factores de riesgo para el desarrollo de ONJ incluyen la terapia inmunosupresora, el tratamiento con inhibidores de la angiogénesis, los corticosteroides sistémicos, la diabetes y las infecciones gingivales. De manera similar, para los pacientes con Xgeva con mieloma múltiple que desarrollaron ONJ, el 58% tenía antecedentes de procedimientos dentales invasivos como factor predisponente.
Realice un examen oral y odontología preventiva adecuada antes de iniciar el tratamiento con Xgeva y periódicamente durante el tratamiento con Xgeva. Avise a los pacientes sobre las prácticas de higiene oral. Evite los procedimientos dentales invasivos durante el tratamiento con Xgeva. Considere la posibilidad de suspender temporalmente el tratamiento con Xgeva si se debe realizar un procedimiento dental invasivo. No hay datos disponibles que sugieran la duración óptima de la interrupción del tratamiento.
Los pacientes que se sospecha que tienen o que desarrollan ONJ mientras están en Xgeva deben recibir atención de un dentista o un cirujano oral. En estos pacientes, la cirugía dental extensa para tratar la ONJ puede exacerbar la afección. El juicio clínico del profesional sanitario tratante debe guiar el plan de manejo de cada paciente en función de la evaluación individual de riesgo/beneficio.
5.5 Fractura femoral subtrocantérica y diafisaria atípica
Se ha notificado fractura femoral atípica con Xgeva [see Adverse Reactions (6.1)]. Estas fracturas pueden ocurrir en cualquier parte del eje femoral, desde justo debajo del trocánter menor hasta por encima de la expansión supracondílea, y tienen una orientación transversal u oblicua corta sin evidencia de conminución.
Las fracturas femorales atípicas ocurren con mayor frecuencia con un trauma mínimo o nulo en el área afectada. Pueden ser bilaterales y muchos pacientes informan dolor prodromal en el área afectada, que generalmente se presenta como dolor sordo y punzante en el muslo, semanas o meses antes de que ocurra una fractura completa. Varios informes señalan que los pacientes también estaban recibiendo tratamiento con glucocorticoides (por ejemplo, prednisona) en el momento de la fractura.
Durante el tratamiento con Xgeva, se debe aconsejar a los pacientes que informen cualquier dolor nuevo o inusual en el muslo, la cadera o la ingle. Cualquier paciente que presente dolor en el muslo o la ingle debe sospecharse que tiene una fractura atípica y debe evaluarse para descartar una fractura de fémur incompleta. El paciente que presenta una fractura de fémur atípica también debe evaluarse para detectar síntomas y signos de fractura en la extremidad contralateral. Se debe considerar la interrupción del tratamiento con Xgeva, en espera de una evaluación de riesgo/beneficio, de forma individual.
5.6 Hipercalcemia después de la interrupción del tratamiento en pacientes con tumor de células gigantes del hueso y en pacientes con esqueletos en crecimiento
Se ha informado de hipercalcemia clínicamente significativa que requiere hospitalización y se complica con lesión renal aguda en pacientes tratados con Xgeva con tumor de células gigantes del hueso y pacientes con esqueletos en crecimiento. Se ha informado de hipercalcemia dentro del primer año después de la interrupción del tratamiento. Después de interrumpir el tratamiento, controle a los pacientes para detectar signos y síntomas de hipercalcemia, evalúe el calcio sérico periódicamente, vuelva a evaluar los requisitos de suplementos de calcio y vitamina D del paciente y maneje a los pacientes según sea clínicamente apropiado [ver Reacciones adversas (6) y Uso en poblaciones específicas (8.4)].
5.7 Fracturas vertebrales múltiples (MVF) después de la interrupción del tratamiento
Se han informado fracturas vertebrales múltiples (MVF) después de la interrupción del tratamiento con denosumab. Los pacientes con mayor riesgo de MVF incluyen aquellos con factores de riesgo o antecedentes de osteoporosis o fracturas previas.
Cuando se interrumpe el tratamiento con Xgeva, evalúe el riesgo individual del paciente de fracturas vertebrales [ver Información para el paciente (17)].
5.8 Toxicidad embrionaria y fetal
Con base en los datos de estudios en animales y su mecanismo de acción, Xgeva puede causar daño fetal cuando se administra a una mujer embarazada. En estudios de reproducción en animales, la administración de denosumab a monos cynomolgus durante todo el embarazo a una dosis 25 veces mayor que la dosis humana recomendada de Xgeva en función del peso corporal resultó en un aumento de la pérdida fetal, muertes fetales y mortalidad posnatal, junto con evidencia de ausencia de ganglios linfáticos periféricos, crecimiento óseo anormal y disminución del crecimiento neonatal.
Verifique el estado de embarazo de las mujeres en edad fértil antes de iniciar el tratamiento con Xgeva. Avise a las mujeres embarazadas y a las mujeres en edad fértil que la exposición a Xgeva durante el embarazo o dentro de los 5 meses anteriores a la concepción puede provocar daño fetal. Avise a las mujeres en edad fértil que usen métodos anticonceptivos efectivos durante la terapia y durante al menos 5 meses después de la última dosis de Xgeva [ver Uso en poblaciones específicas (8.1, 8.3) y Farmacología clínica (12.1)].
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas se abordan a continuación y en otros lugares de la etiqueta:
• Hipersensibilidad [ver Advertencias y Precauciones (5.2)]
• Hipocalcemia [ver Advertencias y Precauciones (5.3) y Uso en Poblaciones Específicas (8.6)]
• Osteonecrosis de la mandíbula [ver Advertencias y Precauciones (5.4)]
• Fractura femorales subtrocantérea y diafisaria atípicas [ver Advertencias y Precauciones (5.5)]
• Hipercalcemia después de la interrupción del tratamiento en pacientes con tumor de células gigantes óseo y en pacientes con esqueletos en crecimiento [ver Advertencias y Precauciones (5.6) y Uso en Poblaciones Específicas (8.4)]
• Fracturas vertebrales múltiples (MVF) después de la interrupción del tratamiento [ver Advertencias y Precauciones (5.7)]
6.1 Experiencia en Ensayos Clínicos
Debido a que los ensayos clínicos se realizan en condiciones muy variadas, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y puede que no reflejen las tasas observadas en la práctica.
Metástasis óseas de tumores sólidos
La seguridad de Xgeva se evaluó en tres ensayos aleatorizados, doble ciego, doble simulado [ver Ensayos Clínicos (14.1)], en los cuales un total de 2841 pacientes con metástasis óseas de cáncer de próstata, cáncer de mama, u otros tumores sólidos, o lesiones óseas líticas de mieloma múltiple recibieron al menos una dosis de Xgeva. En los Estudios 20050136, 20050244 y 20050103, los pacientes fueron aleatorizados a recibir ya sea 120 mg de Xgeva cada 4 semanas como inyección subcutánea o 4 mg (dosis ajustada por disfunción renal reducida) de ácido zoledrónico cada 4 semanas por infusión intravenosa (IV). Los criterios de entrada incluían calcio sérico (corregido) de 8 a 11,5 mg/dL (2 a 2,9 mmol/L) y aclaramiento de creatinina de 30 mL/min o más. Se excluyeron pacientes que habían recibido bifosfonatos intravenosos, así como pacientes con antecedentes de ONJ u osteomielitis de la mandíbula, una condición dental o de la mandíbula activa que requiera cirugía oral, cirugía dental/oral no cicatrizada o cualquier procedimiento dental invasivo planificado. Durante el estudio, las quimias del suero, incluyendo calcio y fósforo, se monitorizaron cada 4 semanas. Se recomendó pero no se requirió la suplementación de calcio y vitamina D.
La duración mediana de exposición a Xgeva fue de 12 meses (rango: 0,1-41) y la duración mediana en el estudio fue de 13 meses (rango: 0,1-41). De los pacientes que recibieron Xgeva, el 46 % eran mujeres. El 85 % eran blancos, el 5 % hispanos/latinos, el 6 % asiáticos y el 3 % negros. La edad mediana era de 63 años (rango: 18-93). El 75 % de los pacientes que recibieron Xgeva recibieron quimioterapia concomitante.
Las reacciones adversas más comunes en los pacientes (incidencia mayor o igual a 25 %) fueron fatiga/astenia, hipofosfatemia y náuseas (ver Tabla 1). La reacción adversa seria más común fue disnea. Las reacciones adversas más comunes que dieron lugar a la interrupción de Xgeva fueron osteonecrosis e hipocalcemia.
| Sistema Corporal | Xgeva n = 2841 % |
Ácido Zoledrónico n = 2836 % |
| GASTROINTESTINAL | ||
| Náuseas | 31 | 32 |
| Diarrea | 20 | 19 |
| GENERAL | ||
| Fatiga/Astenia | 45 | 46 |
| INVESTIGACIONES | ||
| Hipocalcemiab | 18 | 9 |
| Hipofosfatemiab | 32 | 20 |
| NEUROLÓGICO | ||
| Cefalea | 13 | 14 |
| RESPIRATORIO | ||
| Disnea | 21 | 18 |
| Tos | 15 | 15 |
a Reacciones adversas notificadas en al menos el 10% de los pacientes que recibieron Xgeva en los estudios 20050136, 20050244 y 20050103, y que cumplieron uno de los siguientes criterios:
b Derivado de laboratorio y por debajo del límite inferior del laboratorio central de lo normal [8.3 – 8.5 mg/dL (2.075 – 2.125 mmol/L) para calcio y 2.2 – 2.8 mg/dL (0.71 – 0.9 mmol/L) para fósforo] |
||
Anormalidades graves de minerales/electrolitos
▪ Hipocalcemia grave ( calcio sérico corregido inferior a 7 mg/dL o inferior a 1,75 mmol/L) se produjo en el 3,1 % de los pacientes tratados con Xgeva y en el 1,3 % de los tratados con ácido zoledrónico. De los pacientes que experimentaron hipocalcemia grave, el 33 % experimentó 2 o más episodios de hipocalcemia grave y el 16 % experimentó 3 o más episodios [ver Advertencias y Precauciones (5.3) y Uso en Poblaciones Específicas (8.6)].
▪ Hipofosfatemia grave (fosfato sérico inferior a 2 mg/dL o inferior a 0,6 mmol/L) se produjo en el 15,4 % de los pacientes tratados con Xgeva y en el 7,4 % de los pacientes tratados con ácido zoledrónico.
Osteonecrosis de la mandíbula (ONJ)
En las fases de tratamiento primario de los Estudios 20050136, 20050244 y 20050103, ONJ se confirmó en un 1,8 % de los pacientes del grupo Xgeva (exposición mediana de 12,0 meses; rango: 0,1 – 40,5) y en un 1,3 % de los pacientes del grupo de ácido zoledrónico. Los ensayos en pacientes con cáncer de mama (Estudio 20050136) o de próstata (Estudio 20050103) incluyeron una fase de extensión de tratamiento de apertura de etiqueta de Xgeva en la que se ofreció a los pacientes Xgeva 120 mg una vez cada 4 semanas (exposición global mediana de 14,9 meses; rango: 0,1 – 67,2). La incidencia ajustada por paciente-año (número de eventos por 100 paciente-años) de ONJ confirmada fue del 1,1 % durante el primer año de tratamiento, del 3,7 % en el segundo año y del 4,6 % por año a partir de entonces. El tiempo medio hasta ONJ fue de 20,6 meses (rango: 4 – 53) [ver Advertencias y Precauciones (5.4)].
En un ensayo clínico aleatorizado controlado con placebo con una fase de tratamiento de extensión que evaluaba Xgeva para la prevención de metástasis óseas en pacientes con cáncer de próstata no metastásico (una población de pacientes para la que Xgeva no está indicado), con una exposición más larga al tratamiento de hasta 7 años, la incidencia ajustada por paciente-año (número de eventos por 100 paciente-años) de ONJ confirmada fue del 1,1 % durante el primer año de tratamiento, del 3,0 % en el segundo año y del 7,1 % por año a partir de entonces.
Fractura atípica subtrocantérea y diafisaria
En el programa de ensayos clínicos, se ha notificado fractura femoral atípica en pacientes tratados con Xgeva y el riesgo aumentó con la duración más larga del tratamiento [ver Advertencias y Precauciones (5.5)].
Mieloma múltiple
La seguridad de Xgeva se evaluó en un ensayo clínico internacional, aleatorizado (1: 1), doble ciego, controlado activamente de pacientes con mieloma múltiple de nuevo diagnóstico con tratamiento a través de la progresión de la enfermedad [ver Ensayos Clínicos (14.2)]. En este ensayo, los pacientes recibieron 120 mg de Xgeva cada 4 semanas como inyección subcutánea (n = 850) o 4 mg (dosis ajustada para la función renal) de ácido zoledrónico por vía intravenosa (IV) cada 4 semanas por perfusión IV (n = 852). Los criterios de entrada incluyeron calcio sérico (corregido) de 8 a 11,5 mg/dL (2 a 2,9 mmol/L) y depuración de creatinina de 30 mL/min o más. Los pacientes que habían recibido bisfosfonatos por vía IV estaban excluidos, al igual que los pacientes con antecedentes previos de ONJ u osteomielitis de la mandíbula, una condición dental o mandibular activa que requiriera cirugía oral, cirugía dental/oral no curada o cualquier procedimiento dental invasivo planificado. Durante el estudio, las quimias del suero incluyendo calcio y fósforo se monitorizaron cada 4 semanas. Se recomendó pero no se requirió la suplementación con calcio y vitamina D.
La duración mediana de exposición a Xgeva fue de 16 meses (rango: 1 – 50) y la duración mediana en el estudio fue de 17 meses (rango: 0 – 49). De los pacientes que recibieron Xgeva, el 46 % eran mujeres, el 83 % eran blancos, el 13 % asiáticos, el 3 % negros o afroamericanos y el 4 % hispano/latino. La edad mediana de los pacientes aleatorizados a Xgeva fue de 63 años (rango: 29 – 91) y todos los pacientes que recibieron Xgeva recibieron quimioterapia anti-mieloma concomitante.
El perfil de reacción adversa de Xgeva en pacientes con mieloma múltiple, Estudio 20090482, fue similar al observado en los Estudios 20050136, 20050244 y 20050103. Las reacciones adversas más comunes (incidencia ≥ 10 %) fueron diarrea (34 %), náuseas (32 %), anemia (22 %), dolor de espalda (21 %), trombocitopenia (19 %), edema periférico (17 %), hipocalcemia (16 %), infección de las vías respiratorias superiores (15 %), erupción (14 %) y cefalea (11 %). La reacción adversa grave más común (incidencia ≥ 5 %) fue neumonía (8 %). La reacción adversa más común que resultó en la suspensión de Xgeva (≥ 1,0 %) fue osteonecrosis de la mandíbula.
Hipocalcemia e hipofosfatemia
Hipocalcemia grave (calcio sérico corregido inferior a 7 mg/dL o inferior a 1,75 mmol/L) y hipofosfatemia grave (fosfato sérico inferior a 2 mg/dL o inferior a 0,6 mmol/L) ocurrieron en el 2 % y el 21 % de los pacientes tratados con Xgeva, respectivamente.
Osteonecrosis de la mandíbula (ONJ)
En la fase de tratamiento primario del Estudio 20090482, ONJ se confirmó en el 4,1 % de los pacientes en el grupo Xgeva (exposición mediana de 16 meses; rango: 1 – 50) y en el 2,8 % de los pacientes en el grupo de ácido zoledrónico (mediana de 15 meses, rango: 1 – 45 meses). Al final de la fase de tratamiento doble ciego del Estudio 20090482, la incidencia ajustada por paciente-año (número de eventos por 100 paciente-años) de ONJ confirmada en el grupo Xgeva (exposición mediana de 19,4 meses; rango 1 – 52) fue del 2,0 % durante el primer año de tratamiento, del 5,0 % en el segundo año y del 4,5 % por año a partir de entonces. El tiempo medio hasta ONJ fue de 18,7 meses (rango: 1 – 44) [ver Advertencias y Precauciones (5.4)].
Tumor gigante de células de la médula ósea
La seguridad de Xgeva se evaluó en dos ensayos de un solo brazo (Estudio 20062004 y Estudio 20040215) [ver Ensayos clínicos (14.3)] en los que un total de 548 pacientes adultos o adolescentes esqueléticamente maduros con tumor de células gigantes del hueso recibieron al menos 1 dosis de Xgeva. Los pacientes recibieron 120 mg de Xgeva por vía subcutánea cada 4 semanas con dosis adicionales de 120 mg en los días 8 y 15 del primer mes de terapia. Los pacientes que recibieron terapia concomitante con bisfosfonatos fueron excluidos de la inscripción en ambos estudios. Los pacientes con antecedentes de ONJ u osteomielitis de la mandíbula, una condición dental o de la mandíbula activa que requiera cirugía oral, cirugía dental/oral no cicatrizada o cualquier procedimiento dental invasivo planificado fueron excluidos de la inscripción en el Estudio 20040215. Durante el ensayo, se monitorearon las pruebas químicas del suero, incluido el calcio y el fósforo, cada 4 semanas. Se recomendó la suplementación con calcio y vitamina D, pero no fue obligatoria.
De los 548 pacientes que recibieron Xgeva, 467 pacientes fueron tratados con Xgeva durante ≥ 1 año, 323 pacientes durante ≥ 2 años y 255 pacientes durante ≥ 3 años. El número medio de dosis recibidas fue de 33 (rango: 4-138 dosis) y el número medio de meses en el estudio fue de 60 (rango: 0-140 meses). El cincuenta y siete por ciento de los pacientes inscritos fueron mujeres y el 82% fueron blancos. La edad media fue de 33 años (rango: 13-83 años); un total de 19 pacientes fueron adolescentes esqueléticamente maduros (de 12 a <17 años de edad).
El perfil de reacciones adversas común de Xgeva en pacientes con tumor de células gigantes del hueso fue generalmente similar al informado en los Estudios 20050136, 20050244 y 20050103. Las reacciones adversas más comunes en los pacientes (incidencia ≥ 10%) fueron artralgia, dolor de espalda, dolor en las extremidades, fatiga, dolor de cabeza, náuseas, nasofaringitis, dolor musculoesquelético, dolor de muelas, vómitos, hipofosfatemia, estreñimiento, diarrea y tos. Las reacciones adversas graves más frecuentes fueron osteonecrosis de la mandíbula (3,6%), tumor de células gigantes del hueso (1,5%), anemia (1,1%), neumonía (0,9%) y dolor de espalda (0,9%). La reacción adversa más frecuente que provocó la interrupción de Xgeva fue la osteonecrosis de la mandíbula (incidencia del 3,6%). El perfil de reacciones adversas pareció similar en adolescentes esqueléticamente maduros y adultos.
Hipocalcemia e hipofosfatemia
- La hipocalcemia moderada a grave (calcio sérico corregido inferior a 8 mg/dL o inferior a 2 mmol/L) se produjo en el 5% de los pacientes tratados con Xgeva.
- La hipofosfatemia grave (fósforo sérico inferior a 2 a 1 mg/dL o inferior a 0,6 a 0,3 mmol/L) se produjo en el 20% de los pacientes tratados con Xgeva.
Osteonecrosis de la mandíbula (ONJ)
En el Estudio 20062004 y el Estudio 20040215, la ONJ se confirmó en el 6,6% de los pacientes que recibieron Xgeva. [ver Advertencias y precauciones (5.4)].
Fractura atípica subtrocantérica y diafisaria
Se ha informado de fractura femoral atípica con Xgeva y se observó en el 0,9% de los pacientes en la población de seguridad agrupada [ver Advertencias y precauciones (5.5)].
Hipercalcemia después de la interrupción del tratamiento
En la población de seguridad agrupada, el 0,7% de los pacientes experimentaron eventos adversos graves de hipercalcemia > 30 días después de la interrupción del tratamiento que fue recurrente en algunos pacientes [ver Advertencias y precauciones (5.6)].
Hipercalcemia de la malignidad
Xgeva se evaluó en un ensayo abierto, de un solo brazo (Estudio 20070315) en el que se inscribieron 33 pacientes con hipercalcemia de la malignidad (con o sin metástasis óseas) refractaria al tratamiento con terapia con bisfosfonatos intravenosos [ver Ensayos clínicos (14.4)].
El perfil de reacciones adversas de Xgeva en pacientes con hipercalcemia de la malignidad fue similar al informado en los Estudios 20050136, 20050244, 20050103, 20062004 y 20040215. Las reacciones adversas que ocurrieron en más del 20% de los pacientes fueron náuseas (30%), disnea (27%), disminución del apetito (24%), dolor de cabeza (24%), edema periférico (24%), vómitos (24%), anemia (21%), estreñimiento (21%) y diarrea (21%). Las siguientes reacciones adversas de Grado 3 o mayor gravedad relacionadas con la terapia del estudio se informaron en el estudio: fatiga (3%) e infección (6%). Las anormalidades de laboratorio de Grado 3 incluyeron hipomagnesemia (3%), hipopotasemia (3%) e hipofosfatemia (76%) de los pacientes. Ninguna muerte en el estudio estuvo relacionada con la terapia con Xgeva.
6.2 Experiencia postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de Xgeva. Debido a que estas reacciones se informan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
- Hipocalcemia: Hipocalcemia sintomática grave, incluyendo casos fatales [ver Contraindicaciones (4.1) y Advertencias y precauciones (5.3)].
- Hipercalcemia: Hipercalcemia sintomática grave después de la interrupción del tratamiento puede ocurrir [ver Reacciones adversas (6) y Advertencias y precauciones (5.6)].
- Hipersensibilidad, incluyendo reacciones anafilácticas [ver Contraindicaciones (4.2) y Advertencias y precauciones (5.2)].
- Dolor musculoesquelético, incluyendo dolor musculoesquelético grave. Se ha informado de re-desafío positivo.
- Erupciones medicamentosas liquenoides (por ejemplo, reacciones similares al liquen plano).
- Alopecia.
6.3 Inmunogenicidad
Al igual que con todas las proteínas terapéuticas, existe la posibilidad de inmunogenicidad. La detección de la formación de anticuerpos depende en gran medida de la sensibilidad y especificidad de la prueba. Además, la incidencia observada de positividad de anticuerpos (incluidos los anticuerpos neutralizantes) en una prueba puede verse influenciada por varios factores, incluida la metodología de la prueba, el manejo de la muestra, el momento de la recolección de la muestra, los medicamentos concomitantes y la enfermedad subyacente. Por estas razones, la comparación de la incidencia de anticuerpos a denosumab en los estudios descritos a continuación con la incidencia de anticuerpos a otros estudios o a otros productos puede ser engañosa.
Utilizando un inmunoensayo de puente electroquimioluminiscente, menos del 1% (7/2758) de los pacientes con metástasis óseas tratados con denosumab en dosis que van de 30 a 180 mg cada 4 semanas o cada 12 semanas durante un máximo de 3 años dieron positivo para anticuerpos de unión contra denosumab. Ninguno de los 37 pacientes con tumor de células gigantes del hueso en el Estudio 20040215 dio positivo para anticuerpos de unión contra denosumab. Tres de los 506 pacientes con tumor de células gigantes del hueso en el Estudio 20062004 dieron positivo para anticuerpos de unión transitorios después del tratamiento con denosumab. En pacientes con mieloma múltiple en el Estudio 20090482, 1 de 199 pacientes con un resultado posterior a la línea de base, dio positivo para anticuerpos de unión contra denosumab. Ningún paciente con anticuerpos de unión positivos dio positivo para anticuerpos neutralizantes según lo evaluado mediante un ensayo biológico in vitro basado en células quimioluminiscente. No hubo evidencia de un perfil farmacocinético alterado, un perfil de toxicidad o una respuesta clínica a denosumab asociado con el desarrollo de anticuerpos de unión.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen del riesgo
De acuerdo con los hallazgos en animales y su mecanismo de acción, Xgeva puede causar daño fetal cuando se administra a una mujer embarazada [ver Farmacología clínica (12.1)]. No hay datos suficientes sobre el uso de denosumab en mujeres embarazadas para informar sobre los riesgos asociados a los medicamentos para resultados adversos en el desarrollo. La exposición a denosumab en el útero de monos cynomolgus a los que se les administró denosumab mensualmente durante el embarazo a una dosis 25 veces superior a la dosis humana recomendada de Xgeva en función del peso corporal dio lugar a un aumento de la pérdida fetal, mortinatos y mortalidad postnatal; y ausencia de ganglios linfáticos, crecimiento óseo anormal y disminución del crecimiento neonatal [ver Datos].
Informe a las mujeres embarazadas sobre el riesgo potencial para el feto.
Se desconoce la tasa de antecedentes de defectos congénitos importantes y abortos espontáneos para la población indicada. En la población general de los Estados Unidos, el riesgo de antecedentes estimado de defectos congénitos importantes y abortos espontáneos en embarazos clínicamente reconocidos es del 2-4% y del 15-20%, respectivamente.
Datos
Datos en animales
Los efectos del denosumab sobre el desarrollo prenatal se han estudiado tanto en monos cynomolgus como en ratones modificados genéticamente en los que la expresión del ligando RANKL (RANKL) se desactivó mediante la eliminación de genes (un “ratón knockout”). En monos cynomolgus a los que se les administró denosumab por vía subcutánea durante todo el embarazo a partir del día 20 de gestación y a una dosis farmacológicamente activa 25 veces superior a la dosis humana recomendada de Xgeva en función del peso corporal, se produjo un aumento de la pérdida fetal durante la gestación, mortinatos y mortalidad postnatal. Otros hallazgos en las crías incluyeron ausencia de ganglios linfáticos axilares, inguinales, mandibulares y mesentéricos; crecimiento óseo anormal, reducción de la resistencia ósea, reducción de la hematopoyesis, displasia dental y desalineación de los dientes; y disminución del crecimiento neonatal. Al nacer y hasta el mes de edad, los lactantes tenían niveles sanguíneos medibles de denosumab (22-621% de los niveles maternos).
Tras un periodo de recuperación desde el nacimiento hasta los 6 meses de edad, los efectos sobre la calidad y la resistencia ósea volvieron a la normalidad; no hubo efectos adversos sobre la erupción dental, aunque la displasia dental seguía siendo evidente; los ganglios linfáticos axilares e inguinales seguían ausentes, mientras que los ganglios linfáticos mandibulares y mesentéricos estaban presentes, aunque pequeños; y se observó una mineralización de mínima a moderada en múltiples tejidos en un animal en recuperación. No hubo evidencia de daño materno antes del parto; los efectos adversos maternos ocurrieron con poca frecuencia durante el parto. El desarrollo de las glándulas mamarias maternas fue normal. No se estableció ningún NOAEL (nivel sin efectos adversos observables) fetal para este estudio porque solo se evaluó una dosis de 50 mg/kg. La histopatología de las glándulas mamarias a los 6 meses de edad fue normal en las crías hembras expuestas a denosumab en el útero; sin embargo, el desarrollo y la lactancia no se han evaluado completamente.
En ratones knockout para RANKL, la ausencia de RANKL (el objetivo del denosumab) también causó agenesia de los ganglios linfáticos fetales y condujo a un deterioro postnatal de la dentición y el crecimiento óseo. Las ratonas knockout para RANKL preñadas mostraron una maduración alterada de la glándula mamaria materna, lo que condujo a una lactancia deficiente [ver Uso en poblaciones específicas (8.3) y Toxicología no clínica (13.2)].
8.2 Lactancia
Resumen del riesgo
No hay información sobre la presencia de Xgeva (denosumab) en la leche humana, los efectos en el lactante o los efectos en la producción de leche. Se detectó denosumab en la leche materna de monos cynomolgus hasta 1 mes después de la última dosis de denosumab (relación leche:suero ≤ 0.5%) y el desarrollo de las glándulas mamarias maternas fue normal, sin deterioro de la lactancia. Sin embargo, las ratonas knockout para RANKL preñadas mostraron una maduración alterada de la glándula mamaria materna, lo que condujo a una lactancia deficiente [ver Uso en poblaciones específicas (8.1) y Toxicología no clínica (13.2)]. Considere los beneficios para el desarrollo y la salud de la lactancia materna junto con la necesidad clínica de la madre de recibir tratamiento con Xgeva y cualquier efecto adverso potencial sobre el lactante por Xgeva o por la condición materna subyacente.
8.3 Mujeres y hombres con potencial reproductivo
De acuerdo con los hallazgos en animales y su mecanismo de acción, Xgeva puede causar daño fetal cuando se administra a una mujer embarazada [ver Uso en poblaciones específicas (8.1)].
Prueba de embarazo
Verifique el estado de embarazo de las mujeres con potencial reproductivo antes de iniciar el tratamiento con Xgeva.
Anticoncepción
Mujeres
Aconseje a las mujeres con potencial reproductivo que utilicen un método anticonceptivo eficaz durante el tratamiento y durante al menos 5 meses después de la última dosis de Xgeva.
8.4 Uso pediátrico
No se ha establecido la seguridad y eficacia de Xgeva en pacientes pediátricos, excepto en adolescentes con esqueleto maduro (de 12 a 16 años de edad) con tumor óseo de células gigantes. Xgeva se recomienda solo para el tratamiento de adolescentes con esqueleto maduro (de 12 a 16 años de edad) con tumor óseo de células gigantes [ver Indicaciones y uso (1.2)]. Se ha notificado hipercalcemia clínicamente significativa después de la interrupción del tratamiento en pacientes pediátricos con esqueletos en crecimiento que recibieron denosumab para el tumor óseo de células gigantes o para indicaciones no aprobadas [ver Reacciones adversas (6.2) y Advertencias y precauciones (5.6)].
Se estudió Xgeva en un ensayo abierto que incluyó a un subconjunto de 19 pacientes adolescentes (de 12 a 16 años de edad) con tumor óseo de células gigantes que habían alcanzado la madurez esquelética, definida por al menos 1 hueso largo maduro (p. ej., placa de crecimiento epifisaria cerrada del húmero) y que tenían un peso corporal ≥ 45 kg [ver Indicaciones y uso (1.2) y Ensayos clínicos (14.3)]. Un total de uno de cada cinco (20 %) pacientes adolescentes evaluables tuvo una respuesta objetiva mediante la evaluación independiente retrospectiva de la respuesta radiográfica según los Criterios de evaluación de respuesta en tumores sólidos modificados (RECIST 1.1). El perfil de reacciones adversas y los resultados de eficacia parecieron ser similares en adolescentes y adultos con madurez esquelética [ver Reacciones adversas (6.1) y Ensayos clínicos (14.3)].
Datos en animales
El tratamiento con Xgeva puede afectar el crecimiento óseo en niños con placas de crecimiento abiertas y puede inhibir la erupción de la dentición. En ratas neonatales, la inhibición de RANKL (el objetivo de la terapia con Xgeva) con una construcción de osteoprotegerina unida a Fc (OPG-Fc) en dosis ≤ 10 mg/kg se asoció con la inhibición del crecimiento óseo y la erupción dental. Los primates adolescentes tratados con denosumab en dosis 5 y 25 veces (dosis de 10 y 50 mg/kg) más altas que la dosis humana recomendada de 120 mg administrada una vez cada 4 semanas, según el peso corporal (mg/kg), tuvieron placas de crecimiento anormales, lo que se consideró consistente con la actividad farmacológica de denosumab.
Los monos cynomolgus expuestos in utero a denosumab exhibieron anomalías óseas, hematopoyesis reducida, desalineación de los dientes, crecimiento neonatal disminuido y ausencia de ganglios linfáticos axilares, inguinales, mandibulares y mesentéricos. Algunas anomalías óseas se recuperaron una vez que se suspendió la exposición después del nacimiento; sin embargo, los ganglios linfáticos axilares e inguinales permanecieron ausentes 6 meses después del nacimiento [ver Uso en poblaciones específicas (8.1)].
8.5 Uso geriátrico
Del número total de pacientes en estudios clínicos que recibieron Xgeva (n = 2841) en los Estudios 20050136, 20050244 y 20050103, 1271 (44 %) tenían ≥ 65 años, mientras que 473 pacientes (17 %) tenían ≥ 75 años. De los 859 pacientes en el Estudio 20090482 que recibieron Xgeva, 387 pacientes (45 %) tenían ≥ 65 años, mientras que 141 pacientes (16 %) tenían ≥ 75 años. No se observaron diferencias generales en la seguridad o eficacia entre pacientes mayores y jóvenes.
8.6 Insuficiencia renal
Se realizaron dos ensayos clínicos en pacientes sin cáncer y con diversos grados de función renal.
En un estudio, los pacientes (N = 55) con diversos grados de función renal (que van desde la normalidad hasta la enfermedad renal en etapa terminal que requiere diálisis) recibieron una dosis única de 60 mg de denosumab por vía subcutánea. En un segundo estudio, los pacientes (N = 32) con disfunción renal grave (aclaramiento de creatinina inferior a 30 ml/min o en diálisis) recibieron dos dosis de 120 mg de denosumab por vía subcutánea. En ambos estudios, se observó un mayor riesgo de desarrollar hipocalcemia al aumentar la insuficiencia renal y con una suplementación inadecuada/nula de calcio. La hipocalcemia fue de leve a moderada en el 96 % de los pacientes. Controle los niveles de calcio y la ingesta de calcio y vitamina D [ver Advertencias y precauciones (5.3), Reacciones adversas (6.1) y Farmacología clínica (12.3)].
10 SOBREDOSIS
No hay experiencia con sobredosis de Xgeva.
11 DESCRIPCIÓN
Xgeva (denosumab) es un anticuerpo monoclonal humano IgG2 que se une a RANKL humano. Denosumab tiene un peso molecular aproximado de 147 kDa y se produce en células de mamíferos (ovario de hámster chino) genéticamente modificadas.
Xgeva es una solución estéril, sin conservantes, transparente, incolora a amarillo pálido.
Cada vial de dosis única de Xgeva contiene 120 mg de denosumab, acetato (18 mM), polisorbato 20 (0,01%), sorbitol (4,6%), Agua para inyección (USP) y hidróxido de sodio a un pH de 5,2.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Xgeva se une a RANKL, una proteína transmembrana o soluble esencial para la formación, función y supervivencia de los osteoclastos, las células responsables de la resorción ósea, modulando así la liberación de calcio del hueso. El aumento de la actividad de los osteoclastos, estimulada por RANKL, es un mediador de la patología ósea en tumores sólidos con metástasis óseas. De manera similar, los tumores de células gigantes del hueso consisten en células estromales que expresan RANKL y células gigantes similares a osteoclastos que expresan el receptor RANK, y la señalización a través del receptor RANK contribuye a la osteólisis y al crecimiento tumoral. Xgeva evita que RANKL active su receptor, RANK, en la superficie de los osteoclastos, sus precursores y las células gigantes similares a osteoclastos.
12.2 Farmacodinamia
En pacientes con cáncer de mama y metástasis óseas, la reducción mediana en uNTx/Cr fue del 82% dentro de la 1 semana siguiente al inicio de Xgeva 120 mg administrado por vía subcutánea. En los estudios 20050136, 20050244 y 20050103, la reducción mediana en uNTx/Cr desde el inicio hasta el mes 3 fue aproximadamente del 80% en 2075 pacientes tratados con Xgeva.
En un estudio de fase 3 de pacientes con mieloma múltiple de nuevo diagnóstico que recibieron dosis subcutáneas de Xgeva 120 mg cada 4 semanas (Q4W), se observaron reducciones medianas en uNTx/Cr de aproximadamente el 75% para la semana 5. Las reducciones en los marcadores de recambio óseo se mantuvieron, con reducciones medianas del 74% al 79% para uNTx/Cr desde las semanas 9 a 49 de la dosificación continua de 120 mg Q4W.
En pacientes adultos y adolescentes esqueléticamente maduros con tumor de células gigantes del hueso que recibieron dosis subcutáneas de Xgeva 120 mg Q4W con una dosis de carga de 120 mg en los días 8 y 15, las reducciones medianas en uNTx/Cr desde el inicio fueron del 84% en la semana 13 y del 82% en la semana 25.
12.3 Farmacocinética
Después de la administración subcutánea, la biodisponibilidad fue del 62%. Denosumab mostró una farmacocinética no lineal a dosis inferiores a 60 mg, pero aumentos aproximadamente proporcionales a la dosis en la exposición a dosis más altas.
Con dosis subcutáneas múltiples de 120 mg una vez cada 4 semanas, se observó una acumulación de hasta 2,8 veces en las concentraciones séricas de denosumab y se alcanzó el estado estacionario a los 6 meses. Se logró una concentración media (± desviación estándar) en estado estacionario en suero de 20,5 (± 13,5) mcg/mL a los 6 meses. La vida media de eliminación media fue de 28 días.
En pacientes con mieloma múltiple de nuevo diagnóstico que recibieron 120 mg cada 4 semanas, las concentraciones de denosumab parecen alcanzar el estado estacionario al mes 6. En pacientes con tumor de células gigantes del hueso, después de la administración de dosis subcutáneas de 120 mg una vez cada 4 semanas con dosis adicionales de 120 mg en los días 8 y 15 del primer mes de terapia, las concentraciones séricas medias (± desviación estándar) en suero en el día 8, 15 y un mes después de la primera dosis fueron 19,0 (± 24,1), 31,6 (± 27,3), 36,4 (± 20,6) mcg/mL, respectivamente. Se alcanzó el estado estacionario a los 3 meses después del inicio del tratamiento con una concentración media en suero en estado estacionario de 23,4 (± 12,1) mcg/mL.
Poblaciones especiales
Peso corporal: Se realizó un análisis farmacocinético poblacional para evaluar los efectos de las características demográficas. La depuración y el volumen de distribución de denosumab fueron proporcionales al peso corporal. La exposición en estado estacionario después de la administración subcutánea repetida de 120 mg cada 4 semanas a sujetos de 45 kg y 120 kg fue, respectivamente, un 48% más alta y un 46% más baja que la exposición del sujeto típico de 66 kg.
Edad, sexo y raza: La farmacocinética de denosumab no se vio afectada por la edad, el sexo y la raza.
Pediatría: En pacientes adolescentes esqueléticamente maduros (de 12 a 16 años de edad) con tumor de células gigantes del hueso (GCTB) que recibieron 120 mg cada 4 semanas con una dosis de carga de 120 mg en los días 8 y 15, la farmacocinética de denosumab fue comparable a la observada en pacientes adultos con GCTB.
Insuficiencia hepática: No se han realizado ensayos clínicos para evaluar el efecto de la insuficiencia hepática en la farmacocinética de denosumab.
Insuficiencia renal: En ensayos clínicos de 87 pacientes con diversos grados de disfunción renal, incluidos pacientes en diálisis, el grado de insuficiencia renal no tuvo ningún efecto en la farmacocinética y farmacodinamia de denosumab [ver Uso en poblaciones específicas (8.6)].
Interacciones medicamentosas
No se han realizado ensayos formales de interacción fármaco-fármaco con Xgeva. No hubo evidencia de que varios tratamientos anticancerígenos afectaran la exposición sistémica y el efecto farmacodinámico de denosumab. Las concentraciones séricas de denosumab a los 1 y 3 meses y las reducciones en el marcador de recambio óseo uNTx/Cr (telopéptido N-terminal urinario corregido para la creatinina) a los 3 meses fueron similares en pacientes con y sin terapia previa con bisfosfonatos intravenosos y no se vieron alteradas por la quimioterapia concomitante y/o la terapia hormonal.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
No se ha evaluado el potencial carcinogénico de denosumab en estudios a largo plazo en animales. No se ha evaluado el potencial genotóxico de denosumab.
Denosumab no tuvo ningún efecto sobre la fertilidad femenina ni en los órganos reproductores masculinos en monos a dosis que fueron de 6,5 a 25 veces más altas que la dosis humana recomendada de 120 mg administrada por vía subcutánea una vez cada 4 semanas, según el peso corporal (mg/kg).
13.2 Toxicología y/o Farmacología Animal
Denosumab es un inhibidor de la resorción ósea osteoclástica a través de la inhibición de RANKL.
Debido a que la actividad biológica de denosumab en animales es específica de los primates no humanos, la evaluación de ratones genéticamente modificados (knockout) o el uso de otros inhibidores biológicos de la vía RANK/RANKL, OPG-Fc y RANK-Fc, proporcionó información adicional sobre las propiedades farmacodinámicas de denosumab. Los ratones knockout RANK/RANKL exhibieron ausencia de formación de ganglios linfáticos, así como ausencia de lactancia debido a la inhibición de la maduración de la glándula mamaria (desarrollo de la glándula lobuloalveolar durante el embarazo). Los ratones neonatales knockout RANK/RANKL exhibieron un crecimiento óseo reducido y falta de erupción dental. Un estudio corroborativo en ratas de 2 semanas de edad a las que se administró el inhibidor de RANKL OPG-Fc también mostró un crecimiento óseo reducido, placas de crecimiento alteradas y erupción dental deteriorada. Estos cambios fueron parcialmente reversibles en este modelo cuando se suspendió la dosificación con los inhibidores de RANKL.
14 ENSAYOS CLÍNICOS
14.1 Metástasis ósea de tumores sólidos
La seguridad y eficacia de Xgeva para la prevención de eventos esqueléticos relacionados en pacientes con metástasis ósea de tumores sólidos se demostró en tres ensayos internacionales, aleatorizados (1:1), doble ciego, controlados con activo, de no inferioridad que compararon Xgeva con ácido zoledrónico. En los tres ensayos, los pacientes fueron aleatorizados para recibir 120 mg de Xgeva por vía subcutánea cada 4 semanas o 4 mg de ácido zoledrónico por vía intravenosa (IV) cada 4 semanas (dosis ajustada para la función renal reducida). Los pacientes con aclaramiento de creatinina inferior a 30 mL/min fueron excluidos. En cada ensayo, la principal medida de resultado fue la demostración de no inferioridad del tiempo hasta el primer evento esquelético relacionado (SRE) en comparación con el ácido zoledrónico. Las medidas de resultado de apoyo fueron la superioridad del tiempo hasta el primer SRE y la superioridad del tiempo hasta el primer y posterior SRE; la prueba de estas medidas de resultado se produjo si la principal medida de resultado fue estadísticamente significativa. Un SRE se definió como cualquiera de los siguientes: fractura patológica, radioterapia ósea, cirugía ósea o compresión de la médula espinal.
El estudio 20050136 (NCT00321464) reclutó a 2046 pacientes con cáncer de mama avanzado y metástasis ósea. La aleatorización se estratificó por antecedentes de SRE previo (sí o no), recepción de quimioterapia dentro de las 6 semanas previas a la aleatorización (sí o no), uso previo de bifosfonatos orales (sí o no) y región (Japón u otros países). El cuarenta por ciento de los pacientes tenían un SRE previo, el 40% recibieron quimioterapia dentro de las 6 semanas previas a la aleatorización, el 5% recibieron bifosfonatos orales previos y el 7% fueron reclutados de Japón. La edad media fue de 57 años, el 80% de los pacientes eran blancos y el 99% de los pacientes eran mujeres. El número medio de dosis administradas fue de 18 para denosumab y 17 para ácido zoledrónico.
El estudio 20050244 (NCT00330759) reclutó a 1776 adultos con tumores sólidos distintos del cáncer de mama y el cáncer de próstata resistente a la castración con metástasis ósea y mieloma múltiple. La aleatorización se estratificó por SRE previo (sí o no), terapia antineoplásica sistémica en el momento de la aleatorización (sí o no) y tipo de tumor (cáncer de pulmón de células no pequeñas, mieloma u otro). El ochenta y siete por ciento estaban recibiendo terapia antineoplásica sistémica en el momento de la aleatorización, el 52% tenía un SRE previo, el 64% de los pacientes eran hombres, el 87% eran blancos y la edad media fue de 60 años. Un total del 40% de los pacientes tenían cáncer de pulmón de células no pequeñas, el 10% tenían mieloma múltiple, el 9% tenían carcinoma de células renales y el 6% tenían cáncer de pulmón de células pequeñas. Otros tipos de tumores comprendieron menos del 5% de la población reclutada. El número medio de dosis administradas fue de 7 tanto para denosumab como para ácido zoledrónico.
El estudio 20050103 (NCT00321620) reclutó a 1901 hombres con cáncer de próstata resistente a la castración y metástasis ósea. La aleatorización se estratificó por SRE previo, nivel de PSA (inferior a 10 ng/mL o 10 ng/mL o superior) y recepción de quimioterapia dentro de las 6 semanas previas a la aleatorización (sí o no). El veintiséis por ciento de los pacientes tenían un SRE previo, el 15% de los pacientes tenían un PSA inferior a 10 ng/mL y el 14% recibieron quimioterapia dentro de las 6 semanas previas a la aleatorización. La edad media fue de 71 años y el 86% de los pacientes eran blancos. El número medio de dosis administradas fue de 13 para denosumab y 11 para ácido zoledrónico.
Xgeva retrasó el tiempo hasta el primer SRE después de la aleatorización en comparación con el ácido zoledrónico en pacientes con cáncer de mama o cáncer de próstata resistente a la castración (CRPC) con metástasis óseas (Tabla 2). En pacientes con metástasis ósea debido a otros tumores sólidos o lesiones líticas debido a mieloma múltiple, Xgeva no fue inferior al ácido zoledrónico en retrasar el tiempo hasta el primer SRE después de la aleatorización.
La supervivencia general y la supervivencia libre de progresión fueron similares entre los brazos en los tres ensayos.
| Estudio 20050136 Cáncer de mama metastásico |
Estudio 20050244 Tumores sólidos metastásicos o mieloma múltiple |
Estudio 20050103 CRPC metastásicoa |
|||||||||
| Xgeva N = 1026 |
Ácido zoledrónico N = 1020 |
Xgeva N = 886 |
Zoledrónico Acid N = 890 |
Xgeva N = 950 |
Ácido zoledrónico N = 951 |
||||||
| Primer SRE del estudio | |||||||||||
| Número de pacientes que tuvieron SRE (%) | 315 (30.7) | 372 (36.5) | 278 (31.4) | 323 (36.3) | 341 (35.9) | 386 (40.6) | |||||
| Componentes del primer SRE | |||||||||||
| Radiación ósea | 82 (8.0) | 119 (11.7) | 119 (13.4) | 144 (16.2) | 177 (18.6) | 203 (21.3) | |||||
| Fractura patológica | 212 (20.7) | 238 (23.3) | 122 (13.8) | 139 (15.6) | 137 (14.4) | 143 (15.0) | |||||
| Cirugía ósea | 12 (1.2) | 8 (0.8) | 13 (1.5) | 19 (2.1) | 1 (0.1) | 4 (0.4) | |||||
| Compresión de la médula espinal | 9 (0.9) | 7 (0.7) | 24 (2.7) | 21 (2.4) | 26 (2.7) | 36 (3.8) | |||||
| Tiempo medio hasta SRE (meses) | NRb | 26.4 | 20.5 | 16.3 | 20.7 | 17.1 | |||||
| Hazard Ratio (IC del 95%) | 0.82 (0.71, 0.95) | 0.84 (0.71, 0.98) | 0.82 (0.71, 0.95) | ||||||||
| Valor p de no inferioridad | < 0.001 | < 0.001 | < 0.001 | ||||||||
| Valor p de superioridadc | 0.010 | 0.060 | 0.008 | ||||||||
| Primer y subsecuente SREd | |||||||||||
| Número medio/paciente | 0.46 | 0.60 | 0.44 | 0.49 | 0.52 | 0.61 | |||||
| Razón de tasas (IC del 95%) | 0.77 (0.66, 0.89) | 0.90 (0.77, 1.04) | 0.82 (0.71, 0.94) | ||||||||
| Superioridad valor pe | 0.001 | 0.145 | 0.009 | ||||||||
| a CRPC = cáncer de próstata resistente a la castración. b NR = no alcanzado. c La prueba de superioridad se realizó solo después de que denosumab demostró ser no inferior al ácido zoledrónico dentro del ensayo. d Todos los eventos esqueléticos posteriores a la aleatorización; los eventos nuevos se definieron por la ocurrencia ≥ 21 días después del evento anterior. e Se presentan valores p ajustados. |
|||||||||||
14.2 Mieloma Múltiple
La eficacia de Xgeva para la prevención de eventos esqueléticos relacionados en pacientes con mieloma múltiple recién diagnosticados con tratamiento hasta la progresión de la enfermedad, se evaluó en el Estudio 20090482 (NCT01345019), un ensayo internacional, aleatorizado (1:1), doble ciego, controlado con activo, de no inferioridad que comparó Xgeva con ácido zoledrónico. En este ensayo, los pacientes fueron aleatorizados para recibir 120 mg de Xgeva por vía subcutánea cada 4 semanas o 4 mg de ácido zoledrónico por vía intravenosa (IV) cada 4 semanas (dosis ajustada para la función renal reducida). Se excluyeron los pacientes con aclaramiento de creatinina inferior a 30 mL/min. En este ensayo, la principal medida de resultado de eficacia fue la no inferioridad del tiempo hasta el primer evento esquelético relacionado (SRE). Las medidas de resultado de eficacia adicionales fueron la superioridad del tiempo hasta el primer SRE, el tiempo hasta el primer y posterior SRE, y la supervivencia general. Un SRE se definió como cualquiera de los siguientes: fractura patológica, radioterapia ósea, cirugía ósea o compresión de la médula espinal.
El Estudio 20090482 incluyó a 1718 pacientes con mieloma múltiple recién diagnosticados con lesiones óseas. La aleatorización se estratificó por antecedentes de SRE previo (sí o no), el agente anti-mieloma que se estaba utilizando/planeaba utilizar en la terapia de primera línea (basada en terapia novedosa o no basada en terapia novedosa [las terapias novedosas incluyen bortezomib, lenalidomida o talidomida]), intención de someterse a trasplante autólogo de PBSC (sí o no), estadio al diagnóstico (Sistema de estadificación internacional I o II o III) y región Japón (sí o no). Al momento de la inscripción en el estudio, el 96% de los pacientes estaban recibiendo o planeaban recibir terapia de primera línea anti-mieloma basada en terapia novedosa, el 55% de los pacientes tenían la intención de someterse a trasplante autólogo de PBSC, el 61% de los pacientes tenían un SRE previo, el 32% estaban en el estadio I de ISS, el 38% estaban en el estadio II de ISS y el 29% estaban en el estadio III de ISS, y el 2% se inscribieron en Japón. La edad media fue de 63 años, el 82% de los pacientes eran blancos y el 46% de los pacientes eran mujeres. El número medio de dosis administradas fue de 16 para Xgeva y 15 para el ácido zoledrónico.
Xgeva no fue inferior al ácido zoledrónico en retrasar el tiempo hasta el primer SRE después de la aleatorización (HR = 0,98, IC del 95%, 0,85-1,14). Los resultados para la supervivencia general (SG) fueron comparables entre los grupos de tratamiento con Xgeva y ácido zoledrónico con una razón de riesgo de 0,90 (IC del 95%: 0,70, 1,16).
| Estudio 20090482 Mieloma Múltiple |
||
| Xgeva N = 859 |
Ácido Zoledrónico N = 859 |
|
| Primer SRE del estudio | ||
| Número de pacientes que tuvieron SRE (%) | 376 (43,8) | 383 (44,6) |
| Componentes del primer SRE | ||
| Radiación ósea | 47 (5,5) | 62 (7,2) |
| Fractura patológica | 342 (39,8) | 338 (39,3) |
| Cirugía ósea | 37 (4,3) | 48 (5,6) |
| Compresión de la médula espinal | 6 (0,7) | 4 (0,5) |
| Tiempo medio hasta el SRE (meses) (IC del 95%) |
22,8 (14,7, NEa) |
24 (16,6, 33,3) |
| Razón de riesgo (IC del 95%) | 0,98 (0,85, 1,14) | |
| a NE = no estimable | ||
14.3 Tumor de células gigantes del hueso
La seguridad y eficacia de Xgeva para el tratamiento del tumor de células gigantes del hueso en adultos o adolescentes esqueléticamente maduros se demostraron en dos ensayos abiertos [Estudio 20040215 (NCT00396279) y Estudio 20062004 (NCT00680992)] que reclutaron pacientes con tumor de células gigantes del hueso histológicamente confirmado y medible que era recurrente, irresecable o para el cual la cirugía planificada probablemente resultaría en una morbilidad grave. Los pacientes recibieron 120 mg de Xgeva por vía subcutánea cada 4 semanas con una dosis de carga en los días 8 y 15 del primer ciclo de terapia. Los pacientes que suspendieron Xgeva luego ingresaron a la fase de seguimiento de seguridad durante un mínimo de 60 meses. Se permitió el retratamiento con Xgeva mientras se encontraba en el seguimiento de seguridad para los pacientes que inicialmente demostraron una respuesta a Xgeva (por ejemplo, en el caso de una enfermedad recurrente).
El estudio 20040215 fue un ensayo de un solo brazo, farmacodinámico y de prueba de concepto realizado en 37 pacientes adultos con tumor de células gigantes del hueso irresecable o recurrente. Los pacientes debían tener un tumor de células gigantes del hueso histológicamente confirmado y evidencia radiológica de enfermedad medible de una tomografía computarizada (TC) o resonancia magnética (RM) obtenida dentro de los 28 días anteriores a la inscripción en el estudio. Los pacientes inscritos en el estudio 20040215 se sometieron a una evaluación por TC o RM del tumor de células gigantes del hueso al inicio y trimestralmente durante el tratamiento con Xgeva.
El estudio 20062004 fue un ensayo de cohortes paralelas, de prueba de concepto y de seguridad realizado en 535 pacientes adultos o adolescentes esqueléticamente maduros con tumor de células gigantes del hueso histológicamente confirmado y evidencia de enfermedad activa medible. El estudio 20062004 reclutó a 19 pacientes que tenían entre 12 y 16 años de edad [ver Uso en poblaciones específicas (8.4)]. Los pacientes se inscribieron en una de las tres cohortes: la cohorte 1 inscribió a 268 pacientes con enfermedad quirúrgicamente insalvable (por ejemplo, sitios de enfermedad sacra o espinal, o metástasis pulmonares); la cohorte 2 inscribió a 252 pacientes con enfermedad quirúrgicamente salvable donde el investigador determinó que la cirugía planificada probablemente resultaría en una morbilidad grave (por ejemplo, resección articular, amputación de extremidades o hemipelvectomía); la cohorte 3 inscribió a 15 pacientes que participaron previamente en el estudio 20040215. Los pacientes se sometieron a una evaluación de imágenes del estado de la enfermedad en intervalos determinados por su médico tratante.
Un análisis retrospectivo intermedio concluido por un comité de revisión independiente evaluó la respuesta objetiva en 187 pacientes inscritos y tratados en el estudio 20040215 y el estudio 20062004 para quienes se dispuso de una evaluación radiográfica al inicio y al menos una posterior al inicio (27 de 37 pacientes inscritos en el estudio 20040215 y 160 de 270 pacientes inscritos en las cohortes 1 y 2 del estudio 20062004). La medida de resultado de eficacia primaria fue la tasa de respuesta objetiva utilizando los Criterios de evaluación de respuesta en tumores sólidos (RECIST) v 1.1.
La tasa de respuesta objetiva general (RECIST 1.1) fue del 25% (IC del 95%: 19, 32). Todas las respuestas fueron respuestas parciales. El tiempo medio estimado hasta la respuesta fue de 3 meses. En los 47 pacientes con una respuesta objetiva, la duración media del seguimiento fue de 20 meses (rango: 2-44 meses), y el 51% (24/47) tuvo una duración de la respuesta de al menos 8 meses. Tres pacientes experimentaron progresión de la enfermedad después de una respuesta objetiva.
14.4 Hipercalcemia de malignidad
La seguridad y eficacia de Xgeva se demostraron en un ensayo abierto, de un solo brazo [Estudio 20070315 (NCT00896454)] que reclutó a 33 pacientes con hipercalcemia de malignidad (con o sin metástasis óseas) refractaria al tratamiento con terapia con bisfosfonatos intravenosos. Los pacientes recibieron Xgeva por vía subcutánea cada 4 semanas con dosis adicionales de 120 mg en los días 8 y 15 del primer mes de terapia.
En este ensayo, la hipercalcemia de malignidad refractaria se definió como un calcio corregido por albúmina de > 12.5 mg/dL (3.1 mmol/L) a pesar del tratamiento con terapia con bisfosfonatos intravenosos en 7-30 días antes del inicio de la terapia con Xgeva. La medida de resultado primaria fue la proporción de pacientes que lograron una respuesta, definida como calcio sérico corregido (CSC) ≤ 11.5 mg/dL (2.9 mmol/L), dentro de los 10 días posteriores a la administración de Xgeva. Los datos de eficacia se resumen en la Figura 1 y la Tabla 4. La quimioterapia concomitante no pareció afectar la respuesta a Xgeva.
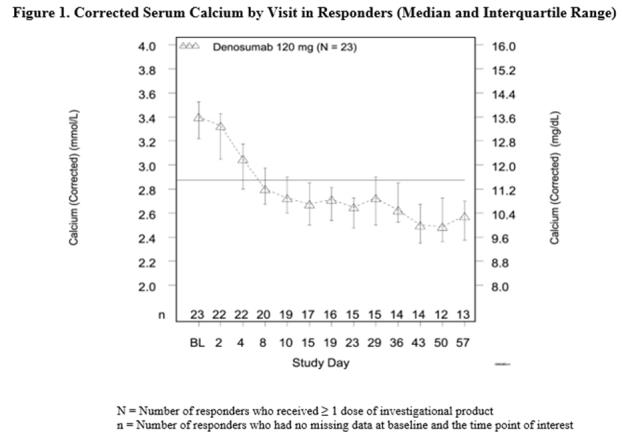
| N = 33 | Proporción (%) (95% CI) |
|
| Todos los respondedores (CSC ≤ 11.5 mg/dL) para el día 10 | 21 | 63.6 |
| (45.1, 79.6) | ||
| Todos los respondedores para el día 57 | 23 | 69.7 |
| (51.3, 84.4) | ||
| Respondedores completos (CSC ≤ 10.8 mg/dL) para el día 10 | 12 | 36.4 |
| (20.4, 54.9) | ||
| Todos los respondedores completos para el día 57 | 21 | 63.6 |
| (45.1, 79.6) |
El tiempo medio hasta la respuesta (CSC ≤ 11.5 mg/dL) fue de 9 días (IC del 95%: 8, 19), y la duración media de la respuesta fue de 104 días (IC del 95%: 7, no estimable). El tiempo medio hasta la respuesta completa (CSC ≤ 10.8 mg/dL) fue de 23 días (IC del 95%: 9, 36), y la duración media de la respuesta completa fue de 34 días (IC del 95%: 1, 134).
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Xgeva se suministra en un vial de dosis única.
| 120 mg/1.7 mL | 1 vial por caja | NDC 55513-730-01 |
Guarde Xgeva en un refrigerador a una temperatura de 2°C a 8°C (36°F a 46°F) en la caja original. No congele. Una vez retirado del refrigerador, Xgeva no debe exponerse a temperaturas superiores a 25°C/77°F ni a luz directa y debe usarse en un plazo de 14 días. Deseche Xgeva si no se utiliza en 14 días. No use Xgeva después de la fecha de caducidad impresa en la etiqueta.
Protéjase Xgeva de la luz directa y el calor.
Evite sacudir enérgicamente Xgeva.
17 INFORMACIÓN PARA EL PACIENTE
Productos farmacéuticos con el mismo ingrediente activo
Avise a los pacientes que el denosumab también se comercializa como Prolia, y si están tomando Xgeva, no deben recibir Prolia [ver Advertencias y precauciones (5.1)].
Hipersensibilidad
Avise a los pacientes que busquen atención médica inmediata si se presentan signos o síntomas de reacciones de hipersensibilidad. Avise a los pacientes que han tenido signos o síntomas de reacciones de hipersensibilidad sistémica que no deben recibir denosumab (Xgeva o Prolia) [ver Advertencias y precauciones (5.2) y Contraindicaciones (4.2)].
Hipocalcemia
Suplemente adecuadamente a los pacientes con calcio y vitamina D e instrúyalos sobre la importancia de mantener los niveles de calcio en suero mientras reciben Xgeva [ver Advertencias y precauciones (5.3) y Uso en poblaciones específicas (8.6)]. Avise a los pacientes que busquen atención médica inmediata si desarrollan signos o síntomas de hipocalcemia.
Osteonecrosis de la mandíbula
Avise a los pacientes que mantengan una buena higiene bucal durante el tratamiento con Xgeva y que informen a su dentista antes de los procedimientos dentales que están recibiendo Xgeva. Los pacientes deben evitar los procedimientos dentales invasivos durante el tratamiento con Xgeva e informar a su proveedor de atención médica o dentista si experimentan dolor persistente y/o curación lenta de la boca o la mandíbula después de la cirugía dental [ver Advertencias y precauciones (5.4)].
Fractura femoral atípica subtrocantérica y diafisaria
Avise a los pacientes que informen cualquier dolor nuevo o inusual en el muslo, la cadera o la ingle [ver Advertencias y precauciones (5.5)].
Hipercalcemia después de la interrupción del tratamiento en pacientes con tumor de células gigantes del hueso y en pacientes con esqueletos en crecimiento
Avise a los pacientes que informen náuseas, vómitos, dolor de cabeza y disminución del estado de alerta después de la interrupción del tratamiento [ver Advertencias y precauciones (5.6) y Uso en poblaciones específicas (8.4)].
Fracturas vertebrales múltiples (MVF) después de la interrupción del tratamiento
Avise a los pacientes que después de detener el tratamiento con Xgeva puede haber un mayor riesgo de fracturas óseas en la columna vertebral, especialmente en pacientes que han tenido una fractura o que han tenido osteoporosis. Avise a los pacientes que no interrumpan el tratamiento con Xgeva sin el consejo de su médico [ver Advertencias y precauciones (5.7)].
Toxicidad embrionaria y fetal
Avise a las mujeres en edad fértil que Xgeva puede causar daño al feto y que informen a su proveedor de atención médica sobre un embarazo conocido o sospechado [ver Advertencias y precauciones (5.8) y Uso en poblaciones específicas (8.1, 8.3)].
Avise a las mujeres en edad fértil que usen métodos anticonceptivos efectivos durante el tratamiento y durante al menos 5 meses después de la última dosis de Xgeva [ver Uso en poblaciones específicas (8.3)].
Xgeva® (denosumab)
Fabricado por:
Amgen Inc.
One Amgen Center Drive
Thousand Oaks, California 91320-1799
Licencia de EE. UU. No. 1080
Patente: http://pat.amgen.com/xgeva/
© 2010-2020 Amgen Inc. Todos los derechos reservados.
1xxxxx – v20
PANEL PRINCIPAL DE VISUALIZACIÓN – Vial de 1.7 mL en caja
AMGEN®
120
mg/1,7 mL
XGEVA®
(denosumab)
120 mg/1,7 mL
(70 mg/mL)
Inyección
Para uso subcutáneo solamente
Envase con vial de dosis única. Deseche la parte no utilizada.
