
Fabricante de medicamentos: Celgene Corporation (Updated: 2023-03-24)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
POMALYST® (pomalidomida) cápsulas, para uso oral
Aprobación inicial en EE. UU.: 2013
ADVERTENCIA: TOXICIDAD EMBRIOFETAL Y TROMBOEMBOLISMO VENOSO Y ARTERIAL
Consulte la información completa de prescripción para la advertencia completa en recuadro
TOXICIDAD EMBRIOFETAL
- •
- POMALYST está contraindicado en el embarazo. POMALYST es un análogo de la talidomida. La talidomida es un teratógeno humano conocido que causa defectos de nacimiento graves que ponen en peligro la vida (4, 5.1, 8.1).
- •
- Para mujeres en edad fértil: Excluir el embarazo antes de comenzar el tratamiento. Evite el embarazo durante el tratamiento mediante el uso de 2 métodos anticonceptivos confiables (5.1, 8.3).
POMALYST solo está disponible a través de un programa restringido llamado POMALYST REMS® (5.2).
TROMBOEMBOLISMO VENOSO Y ARTERIAL
- •
- Trombosis venosa profunda (TVP), embolia pulmonar (EP), infarto de miocardio y accidente cerebrovascular ocurren en pacientes con mieloma múltiple tratados con POMALYST. Se recomienda la profilaxis antitrombótica (5.3).
INDICACIONES Y USO
POMALYST es un análogo de la talidomida indicado para el tratamiento de pacientes adultos:
- •
- en combinación con dexametasona, para pacientes con mieloma múltiple (MM) que han recibido al menos dos terapias previas, incluida la lenalidomida y un inhibidor del proteasoma, y han demostrado progresión de la enfermedad en o dentro de los 60 días posteriores a la finalización de la última terapia (1.1).
- •
- con sarcoma de Kaposi (KS) relacionado con el SIDA después del fracaso de la terapia antirretroviral altamente activa (TARHA) o en pacientes con KS que son VIH negativos. Esta indicación está aprobada bajo aprobación acelerada en función de la tasa de respuesta general. La aprobación continua para esta indicación puede estar supeditada a la verificación y descripción del beneficio clínico en un ensayo confirmatorio (1.2).
DOSIFICACIÓN Y ADMINISTRACIÓN
- •
- MM: 4 mg por día administrados por vía oral los días 1 a 21 de ciclos repetidos de 28 días hasta la progresión de la enfermedad (2.2). Consulte la sección 14.1 para la dosificación de dexametasona (14.1)
- •
- KS: 5 mg por día administrados por vía oral los días 1 a 21 de ciclos repetidos de 28 días hasta la progresión de la enfermedad o toxicidad inaceptable (2.3).
- •
- Modifique la dosis para ciertos pacientes con insuficiencia renal (2.7, 8.6) o insuficiencia hepática (2.8, 8.7)
FORMAS Y FUERZAS DE DOSIFICACIÓN
Cápsulas: 1 mg, 2 mg, 3 mg y 4 mg (3)
ADVERTENCIAS Y PRECAUCIONES
- •
- Aumento de la mortalidad: Observado en pacientes con MM cuando se agregó pembrolizumab a la dexametasona y un análogo de la talidomida (5.4).
- •
- Toxicidad hematológica: La neutropenia fue el evento adverso de Grado 3/4 más comúnmente reportado. Monitoree a los pacientes para detectar toxicidades hematológicas, especialmente neutropenia (5.5).
- •
- Hepatotoxicidad: Insuficiencia hepática, incluidas las muertes; monitorear las pruebas de función hepática mensualmente (5.6).
- •
- Reacciones cutáneas graves: Suspenda POMALYST para reacciones graves (5.7).
- •
- Síndrome de lisis tumoral (SLT): Monitoree a los pacientes en riesgo de SLT (es decir, aquellos con alta carga tumoral) y tome las precauciones apropiadas (5.11).
- •
- Hipersensibilidad: Monitoree a los pacientes para detectar posibles hipersensibilidades. Suspenda POMALYST para angioedema y anafilaxia (5.12).
REACCIONES ADVERSAS
- •
- MM: Las reacciones adversas más comunes (≥30%) incluyeron fatiga y astenia, neutropenia, anemia, estreñimiento, náuseas, diarrea, disnea, infecciones del tracto respiratorio superior, dolor de espalda y pirexia (6.1).
- •
- KS: Las reacciones adversas más comunes, incluidas las anomalías de laboratorio (≥30%), son disminución del recuento absoluto de neutrófilos o glóbulos blancos, aumento de la creatinina o la glucosa, erupción cutánea, estreñimiento, fatiga, disminución de la hemoglobina, plaquetas, fosfato, albúmina o calcio, aumento de la ALT, náuseas y diarrea (6.1).
Para reportar REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Bristol Myers Squibb al 1-800-721-5072 o la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
INTERACCIONES MEDICAMENTOSAS
Inhibidores fuertes de CYP1A2: Evite el uso concomitante de inhibidores fuertes de CYP1A2. Si el uso concomitante de un inhibidor fuerte de CYP1A2 es inevitable, reduzca la dosis de POMALYST a 2 mg (2.6, 7.1, 12.3).
Consulte 17 para obtener INFORMACIÓN PARA EL PACIENTE y la Guía de medicamentos.
Revisado: 3/2023
Tabla de Contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
ADVERTENCIA: TOXICIDAD EMBRIOFETAL Y TROMBOEMBOLISMO VENOSO Y ARTERIAL
1 INDICACIONES Y USO
1.1 Mieloma Múltiple
1.2 Sarcoma de Kaposi
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Prueba de Embarazo Antes de la Administración
2.2 Dosis Recomendada para Mieloma Múltiple
2.3 Dosis Recomendada para Sarcoma de Kaposi
2.4 Modificaciones de la Dosis para Reacciones Adversas Hematológicas
2.5 Modificaciones de la Dosis para Reacciones Adversas No Hematológicas
2.6 Modificaciones de la Dosis para Inhibidores Fuertes de CYP1A2
2.7 Modificación de la Dosis para Insuficiencia Renal Grave en Hemodiálisis
2.8 Modificación de la Dosis para Insuficiencia Hepática
2.9 Administración
3 FORMAS Y FUERZAS DE DOSIFICACIÓN
4 CONTRAINDICACIONES
4.1 Embarazo
4.2 Hipersensibilidad
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Toxicidad Embriofetal
5.2 Programa REMS de POMALYST
5.3 Tromboembolismo Venoso y Arterial
5.4 Aumento de la Mortalidad en Pacientes con Mieloma Múltiple Cuando se Agrega Pembrolizumab a un Análogo de Talidomida y Dexametasona
5.5 Toxicidad Hematológica
5.6 Hepatotoxicidad
5.7 Reacciones Cutáneas Graves
5.8 Mareos y Estado Confusional
5.9 Neuropatía
5.10 Riesgo de Segundas Neoplasias Malignas
5.11 Síndrome de Lisis Tumoral
5.12 Hipersensibilidad
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos
6.2 Experiencia Postcomercialización
7 INTERACCIONES MEDICAMENTOSAS
7.1 Medicamentos que Afectan las Concentraciones Plasmáticas de Pomalidomida
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y Hombres en Potencial Reproductivo
8.4 Uso Pediátrico
8.5 Uso Geriátrico
8.6 Insuficiencia Renal
8.7 Insuficiencia Hepática
8.8 Fumar Tabaco
10 SOBREDOSIS
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
14 ESTUDIOS CLÍNICOS
14.1 Mieloma Múltiple
14.2 Sarcoma de Kaposi
15 REFERENCIAS
16 CÓMO SE SUMINISTRA/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no están enumeradas.
ADVERTENCIA RECUADRADA
ADVERTENCIA: TOXICIDAD EMBRIOFETAL Y TROMBOEMBOLISMO VENOSO Y ARTERIAL
Toxicidad embrio-fetal
- •
- POMALYST está contraindicado en el embarazo. POMALYST es un análogo de la talidomida. La talidomida es un teratógeno humano conocido que causa defectos de nacimiento graves o muerte embrio-fetal. En mujeres en edad fértil, obtenga 2 pruebas de embarazo negativas antes de comenzar el tratamiento con POMALYST.
- •
- Las mujeres en edad fértil deben utilizar 2 métodos de anticoncepción o abstenerse continuamente de las relaciones sexuales heterosexuales durante y durante las 4 semanas posteriores a la interrupción del tratamiento con POMALYST [ver Contraindicaciones (4), Advertencias y precauciones (5.1) y Uso en poblaciones específicas (8.1, 8.3)].
POMALYST solo está disponible a través de un programa de distribución restringido llamado POMALYST REMS [ver Advertencias y precauciones (5.2)]. La información sobre el programa POMALYST REMS está disponible en www.pomalystrems.com o llamando al Centro de llamadas de REMS al 1-888-423-5436.
Tromboembolismo venoso y arterial
- •
- Trombosis venosa profunda (TVP), embolia pulmonar (EP), infarto de miocardio y accidente cerebrovascular ocurren en pacientes con mieloma múltiple tratados con POMALYST. Se emplearon medidas antitrombóticas profilácticas en los ensayos clínicos. Se recomienda la tromboprofilaxis, y la elección del régimen debe basarse en la evaluación de los factores de riesgo subyacentes del paciente [ver Advertencias y precauciones (5.3)].
1 INDICACIONES Y USO
1.1 Mieloma Múltiple
POMALYST, en combinación con dexametasona, está indicado para pacientes adultos con mieloma múltiple (MM) que han recibido al menos dos terapias previas incluyendo lenalidomida y un inhibidor del proteasoma y han demostrado progresión de la enfermedad durante o dentro de los 60 días posteriores a la finalización de la última terapia.
1.2 Sarcoma de Kaposi
POMALYST está indicado para el tratamiento de:
- •
- Pacientes adultos con sarcoma de Kaposi (KS) relacionado con el SIDA después del fracaso de la terapia antirretroviral altamente activa (HAART).
- •
- Sarcoma de Kaposi (KS) en pacientes adultos que son VIH negativos.
Esta indicación está aprobada bajo aprobación acelerada basada en la tasa de respuesta general [ver Estudios Clínicos (14.2)]. La aprobación continua para esta indicación puede estar sujeta a la verificación y descripción del beneficio clínico en un ensayo(s) confirmatorio(s).
2 DOSIS Y ADMINISTRACIÓN
2.1 Prueba de Embarazo Antes de la Administración
Las mujeres en edad fértil deben tener una prueba de embarazo negativa y utilizar métodos anticonceptivos antes de iniciar POMALYST [ver Advertencias y Precauciones (5.1) y Uso en Poblaciones Específicas (8.1, 8.3)].
2.2 Dosis Recomendada para el Mieloma Múltiple
La dosis recomendada de POMALYST es de 4 mg una vez al día por vía oral con o sin alimentos los días 1 a 21 de cada ciclo de 28 días hasta la progresión de la enfermedad. Administre POMALYST en combinación con dexametasona [ver Estudios Clínicos (14.1)].
2.3 Dosis Recomendada para el Sarcoma de Kaposi
La dosis recomendada de POMALYST es de 5 mg una vez al día por vía oral con o sin alimentos los días 1 a 21 de cada ciclo de 28 días hasta la progresión de la enfermedad o la toxicidad inaceptable. Continúe con HAART como tratamiento para el VIH en pacientes con sarcoma de Kaposi (KS) relacionado con el SIDA [ver Estudios Clínicos (14.2)].
2.4 Modificaciones de la Dosis para Reacciones Adversas Hematológicas
Mieloma Múltiple: Modificaciones de la Dosis para Reacciones Adversas Hematológicas
Inicie un nuevo ciclo de POMALYST en pacientes con mieloma múltiple (MM) cuando el recuento de neutrófilos sea al menos 500 por mcL y el recuento de plaquetas sea al menos 50,000 por mcL.
La modificación de la dosis de POMALYST para reacciones adversas hematológicas en pacientes con MM se resume en la Tabla 1.
| Reacción Adversa | Severidad | Modificación de la Dosis |
|---|---|---|
| * Suspenda permanentemente POMALYST si no puede tolerar 1 mg una vez al día. ANC= recuento absoluto de neutrófilos |
||
|
Neutropenia [ver Advertencias y Precauciones (5.5)] |
|
|
|
|
|
|
Trombocitopenia [ver Advertencias y Precauciones (5.5)] |
|
|
|
|
|
Sarcoma de Kaposi: Modificaciones de la dosis para reacciones adversas hematológicas
Inicie un nuevo ciclo de POMALYST en pacientes con KS cuando el recuento de neutrófilos sea al menos 1000 por mcL y el recuento de plaquetas sea al menos 75,000 por mcL.
Las modificaciones de la dosis para POMALYST para reacciones adversas hematológicas en pacientes con KS se resumen en la Tabla 2.
| Reacción adversa | Severidad | Modificación de la dosis |
|---|---|---|
| * Suspenda permanentemente POMALYST si no puede tolerar 1 mg una vez al día. ANC= recuento absoluto de neutrófilos |
||
|
Neutropenia [ver Advertencias y precauciones (5.5)] |
ANC 500 a menos de 1,000 por mcL |
Día 1 del ciclo
Durante el ciclo
|
|
ANC menos de 500 por mcL |
|
|
|
Neutropenia febril [ver Advertencias y precauciones (5.5)] |
ANC menos de 1,000 por mcL y temperatura única mayor o igual a 38.3°C |
|
|
Trombocitopenia [ver Advertencias y precauciones (5.5)] |
Recuento de plaquetas 25,000 a menos de 50,000 por mcL |
Día 1 del ciclo
Durante el ciclo:
|
|
Recuento de plaquetas menos de 25,000 por mcL |
Suspenda permanentemente POMALYST. |
|
2.5 Modificaciones de la Dosis para Reacciones Adversas No Hematológicas
Suspenda permanentemente POMALYST para angioedema, anafilaxia, erupción cutánea de Grado 4, exfoliación cutánea, ampollas o cualquier otra reacción dermatológica grave [Ver Advertencias y Precauciones (5.7, 5.12)].
Para otras toxicidades de Grado 3 o 4, suspenda el tratamiento y reinicie el tratamiento a 1 mg menos que la dosis anterior cuando la toxicidad se haya resuelto a menos que o igual a Grado 2 a discreción del médico.
2.6 Modificaciones de la Dosis para Inhibidores Fuertes de CYP1A2
Evite el uso concomitante de POMALYST con inhibidores fuertes de CYP1A2. Si el uso concomitante de un inhibidor fuerte de CYP1A2 es inevitable, reduzca la dosis de POMALYST a 2 mg [ver Interacciones Medicamentosas (7.1) y Farmacología Clínica (12.3)].
2.7 Modificación de la Dosis para Insuficiencia Renal Grave en Hemodiálisis
Tome POMALYST después de completar el procedimiento de diálisis en los días de hemodiálisis [ver Uso en Poblaciones Específicas (8.6) y Farmacología Clínica (12.3)].
- •
- Para pacientes con MM con insuficiencia renal grave que requieren diálisis, reduzca la dosis recomendada a 3 mg por vía oral al día.
- •
- Para pacientes con KS con insuficiencia renal grave que requieren diálisis, reduzca la dosis recomendada a 4 mg por vía oral al día.
2.8 Modificación de la Dosis para Insuficiencia Hepática
Mieloma Múltiple
Para pacientes con MM con insuficiencia hepática leve o moderada (Child-Pugh A o B), reduzca la dosis recomendada a 3 mg por vía oral al día.
Para pacientes con MM con insuficiencia hepática grave (Child-Pugh C), reduzca la dosis recomendada a 2 mg [ver Uso en Poblaciones Específicas (8.7) y Farmacología Clínica (12.3)].
Sarcoma de Kaposi
Para pacientes con KS con insuficiencia hepática leve, moderada o grave (Child-Pugh A, B o C), reduzca la dosis recomendada a 3 mg por vía oral al día [ver Uso en Poblaciones Específicas (8.7) y Farmacología Clínica (12.3)].
2.9 Administración
Trague las cápsulas enteras con agua. No rompa, mastique ni abra las cápsulas.
POMALYST se puede tomar con o sin alimentos.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
- •
- Cápsulas: 1 mg, tapa opaca azul oscura y cuerpo opaco amarillo, con la impresión “POML” en la tapa en tinta blanca y “1 mg” en el cuerpo en tinta negra
- •
- 2 mg, tapa opaca azul oscura y cuerpo opaco naranja, con la impresión “POML” en la tapa y “2 mg” en el cuerpo en tinta blanca
- •
- 3 mg, tapa opaca azul oscura y cuerpo opaco verde, con la impresión “POML” en la tapa y “3 mg” en el cuerpo en tinta blanca
- •
- 4 mg, tapa opaca azul oscura y cuerpo opaco azul, con la impresión “POML” en la tapa y “4 mg” en el cuerpo en tinta blanca
4 CONTRAINDICACIONES
4.1 Embarazo
POMALYST está contraindicado en mujeres embarazadas. POMALYST puede causar daño fetal cuando se administra a una mujer embarazada [ver Advertencias y precauciones (5.1) y Uso en poblaciones específicas (8.1)]. Pomalidomida es un análogo de la talidomida y es teratógeno tanto en ratas como en conejos cuando se administra durante el período de organogénesis. Si la paciente queda embarazada mientras toma este medicamento, se le debe informar del riesgo potencial para el feto.
4.2 Hipersensibilidad
POMALYST está contraindicado en pacientes que han demostrado hipersensibilidad grave (por ejemplo, angioedema, anafilaxia) a pomalidomida o a cualquiera de los excipientes [ver Advertencias y precauciones (5.7), Descripción (11)].
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Toxicidad embrio-fetal
POMALYST es un análogo de la talidomida y está contraindicado para su uso durante el embarazo. La talidomida es un teratógeno humano conocido que causa defectos de nacimiento graves o muerte embrio-fetal [ver Uso en poblaciones específicas (8.1)]. POMALYST solo está disponible a través del programa POMALYST REMS [ver Advertencias y precauciones (5.2)].
Mujeres en edad fértil
Las mujeres en edad fértil deben evitar el embarazo durante al menos 4 semanas antes de comenzar el tratamiento con POMALYST, durante el tratamiento, durante las interrupciones de la dosis y durante al menos 4 semanas después de completar el tratamiento.
Las mujeres deben comprometerse a abstenerse continuamente de las relaciones sexuales heterosexuales o a utilizar 2 métodos de control de la natalidad fiables, comenzando 4 semanas antes de iniciar el tratamiento con POMALYST, durante el tratamiento, durante las interrupciones de la dosis y continuando durante 4 semanas después de la interrupción del tratamiento con POMALYST.
Se deben obtener dos pruebas de embarazo negativas antes de iniciar el tratamiento. La primera prueba debe realizarse dentro de los 10-14 días y la segunda prueba dentro de las 24 horas previas a la prescripción del tratamiento con POMALYST y luego semanalmente durante el primer mes, luego mensualmente a partir de entonces en mujeres con ciclos menstruales regulares, o cada 2 semanas en mujeres con ciclos menstruales irregulares [ver Uso en poblaciones específicas (8.3)].
Hombres
La pomalidomida está presente en el semen de los pacientes que reciben el medicamento. Por lo tanto, los hombres deben usar siempre un condón de látex o sintético durante cualquier contacto sexual con mujeres en edad fértil mientras toman POMALYST y hasta 4 semanas después de dejar de tomar POMALYST, incluso si se han sometido a una vasectomía exitosa. Los pacientes de sexo masculino que toman POMALYST no deben donar esperma [ver Uso en poblaciones específicas (8.3)].
5.2 Programa POMALYST REMS
Debido al riesgo embrio-fetal [ver Advertencias y precauciones (5.1)], POMALYST solo está disponible a través de un programa restringido bajo una Estrategia de Evaluación y Mitigación de Riesgos (REMS), el programa “POMALYST REMS”.
Los componentes obligatorios del programa POMALYST REMS incluyen los siguientes:
- •
- Los prescriptores deben estar certificados con el programa POMALYST REMS inscribiéndose y cumpliendo con los requisitos de REMS.
- •
- Los pacientes deben firmar un Formulario de Acuerdo Paciente-Médico y cumplir con los requisitos de REMS. En particular, las pacientes de sexo femenino en edad fértil que no están embarazadas deben cumplir con los requisitos de pruebas de embarazo y anticoncepción [ver Uso en poblaciones específicas (8.3)] y los hombres deben cumplir con los requisitos de anticoncepción [ver Uso en poblaciones específicas (8.3)].
- •
- Las farmacias deben estar certificadas con el programa POMALYST REMS, solo deben dispensar a pacientes que están autorizados a recibir POMALYST y cumplir con los requisitos de REMS.
Se puede obtener más información sobre el programa POMALYST REMS en www.pomalystrems.com o por teléfono al 1-888-423-5436.
5.3 Tromboembolismo venoso y arterial
Se han observado eventos tromboembólicos venosos (trombosis venosa profunda y embolia pulmonar) y eventos tromboembólicos arteriales (infarto de miocardio y accidente cerebrovascular) en pacientes tratados con POMALYST. En el ensayo 2, donde se exigieron terapias anticoagulantes, se produjeron eventos tromboembólicos en el 8,0% de los pacientes tratados con POMALYST y dexametasona de dosis baja (dexametasona de dosis baja) y en el 3,3% de los pacientes tratados con dexametasona de dosis alta. Los eventos tromboembólicos venosos (TEV) se produjeron en el 4,7% de los pacientes tratados con POMALYST y dexametasona de dosis baja y en el 1,3% de los pacientes tratados con dexametasona de dosis alta. Los eventos tromboembólicos arteriales incluyen términos para eventos tromboembólicos arteriales, condiciones cerebrovasculares isquémicas y cardiopatía isquémica. Los eventos tromboembólicos arteriales se produjeron en el 3,0% de los pacientes tratados con POMALYST y dexametasona de dosis baja y en el 1,3% de los pacientes tratados con dexametasona de dosis alta.
Los pacientes con factores de riesgo conocidos, incluida la trombosis previa, pueden tener un mayor riesgo, y se deben tomar medidas para intentar minimizar todos los factores modificables (por ejemplo, hiperlipidemia, hipertensión, tabaquismo). Se recomienda la tromboprofilaxis, y la elección del régimen debe basarse en la evaluación de los factores de riesgo subyacentes del paciente.
5.4 Aumento de la mortalidad en pacientes con mieloma múltiple cuando se agrega pembrolizumab a un análogo de la talidomida y dexametasona
En dos ensayos clínicos aleatorizados en pacientes con MM, la adición de pembrolizumab a un análogo de la talidomida más dexametasona, un uso para el cual no está indicado ningún anticuerpo bloqueador de PD-1 o PD-L1, resultó en un aumento de la mortalidad. No se recomienda el tratamiento de pacientes con MM con un anticuerpo bloqueador de PD-1 o PD-L1 en combinación con un análogo de la talidomida más dexametasona fuera de los ensayos clínicos controlados.
5.5 Toxicidad hematológica
Mieloma múltiple
En los ensayos 1 y 2 en pacientes que recibieron POMALYST + dexametasona de dosis baja, la neutropenia fue la reacción adversa de grado 3 o 4 más frecuentemente reportada, seguida de anemia y trombocitopenia. La neutropenia de cualquier grado se reportó en el 51% de los pacientes en ambos ensayos. La tasa de neutropenia de grado 3 o 4 fue del 46%. La tasa de neutropenia febril fue del 8%.
Monitorear a los pacientes para detectar toxicidades hematológicas, especialmente neutropenia. Monitorear los hemogramas completos semanalmente durante las primeras 8 semanas y mensualmente a partir de entonces. Los pacientes pueden requerir interrupción y/o modificación de la dosis [ver Dosificación y administración (2.4)].
Sarcoma de Kaposi
En el ensayo 12-C-0047, las toxicidades hematológicas fueron las reacciones adversas más comunes (todos los grados y grado 3 o 4) [ver Reacciones adversas (6.1)]. El cincuenta por ciento de los pacientes tuvieron neutropenia de grado 3 o 4. Monitorear a los pacientes para detectar toxicidades hematológicas, especialmente disminución de neutrófilos. Monitorear los hemogramas completos cada 2 semanas durante las primeras 12 semanas y mensualmente a partir de entonces. Suspender, reducir la dosis o suspender permanentemente POMALYST según la gravedad de la reacción [ver Dosificación y administración (2.4)].
5.6 Hepatotoxicidad
Se ha producido insuficiencia hepática, incluidos casos mortales, en pacientes tratados con POMALYST. También se han observado niveles elevados de alanina aminotransferasa y bilirrubina en pacientes tratados con POMALYST. Monitorear las pruebas de función hepática mensualmente. Detenga POMALYST en caso de elevación de las enzimas hepáticas y evalúe. Después de volver a los valores basales, se puede considerar el tratamiento a una dosis más baja.
5.7 Reacciones cutáneas graves
Se han notificado reacciones cutáneas graves, incluido el síndrome de Stevens-Johnson (SSJ), la necrólisis epidérmica tóxica (NET) y la reacción medicamentosa con eosinofilia y síntomas sistémicos (DRESS). DRESS puede presentarse con una reacción cutánea (como erupción o dermatitis exfoliativa), eosinofilia, fiebre y/o linfadenopatía con complicaciones sistémicas como hepatitis, nefritis, neumonitis, miocarditis y/o pericarditis. Estas reacciones pueden ser mortales. Considere la interrupción o la suspensión de POMALYST para erupciones cutáneas de grado 2 o 3. Suspenda permanentemente POMALYST para erupciones de grado 4, erupciones exfoliativas o bulbosas, o para otras reacciones cutáneas graves como SSJ, NET o DRESS [ver Dosificación y administración (2.5)].
5.8 Mareos y estado confusional
En los ensayos 1 y 2 en pacientes que recibieron POMALYST + Dex de dosis baja, el 14% de los pacientes experimentaron mareos y el 7% de los pacientes experimentaron un estado confusional; el 1% de los pacientes experimentaron mareos de grado 3 o 4, y el 3% de los pacientes experimentaron un estado confusional de grado 3 o 4. Instruya a los pacientes para que eviten situaciones en las que los mareos o el estado confusional puedan ser un problema y que no tomen otros medicamentos que puedan causar mareos o estado confusional sin el consejo médico adecuado.
5.9 Neuropatía
En los ensayos 1 y 2 en pacientes que recibieron POMALYST + Dex de dosis baja, el 18% de los pacientes experimentaron neuropatía, con aproximadamente el 12% de los pacientes experimentando neuropatía periférica. El dos por ciento de los pacientes experimentaron neuropatía de grado 3 en el ensayo 2. No se reportaron casos de reacciones adversas de neuropatía de grado 4 en ninguno de los ensayos.
5.10 Riesgo de segundas neoplasias malignas primarias
Se han notificado casos de leucemia mieloide aguda en pacientes que recibieron POMALYST como terapia de investigación fuera de MM.
5.11 Síndrome de lisis tumoral
El síndrome de lisis tumoral (SLT) puede ocurrir en pacientes tratados con POMALYST. Los pacientes con riesgo de SLT son aquellos con una alta carga tumoral antes del tratamiento. Estos pacientes deben ser monitoreados de cerca y se deben tomar las precauciones apropiadas.
5.12 Hipersensibilidad
Se ha notificado hipersensibilidad, incluido angioedema, anafilaxia y reacciones anafilácticas a POMALYST. Suspenda permanentemente POMALYST para angioedema o anafilaxia [ver Dosificación y administración (2.5)].
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas clínicamente significativas se describen en detalle en otras secciones del etiquetado:
- •
- Toxicidad embrio-fetal [ver Advertencias y precauciones (5.1, 5.2)]
- •
- Tromboembolismo venoso y arterial [ver Advertencias y precauciones (5.3)]
- •
- Aumento de la mortalidad en pacientes con mieloma múltiple cuando se añade pembrolizumab a un análogo de la talidomida y dexametasona [ver Advertencias y precauciones (5.4)]
- •
- Toxicidad hematológica [ver Advertencias y precauciones (5.5)]
- •
- Hepatotoxicidad [ver Advertencias y precauciones (5.6)]
- •
- Reacciones cutáneas graves [ver Advertencias y precauciones (5.7)]
- •
- Mareos y estado confusional [ver Advertencias y precauciones (5.8)]
- •
- Neuropatía [ver Advertencias y precauciones (5.9)]
- •
- Riesgo de segundas neoplasias malignas [ver Advertencias y precauciones (5.10)]
- •
- Síndrome de lisis tumoral [ver Advertencias y precauciones (5.11)]
- •
- Hipersensibilidad [ver Advertencias y precauciones (5.12)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica.
Mieloma múltiple (MM)
En el ensayo 1, se evaluaron los datos de 219 pacientes (población de seguridad) que recibieron tratamiento con POMALYST + Dex de baja dosis (112 pacientes) o POMALYST solo (107 pacientes). El número medio de ciclos de tratamiento fue de 5. El sesenta y siete por ciento de los pacientes del estudio tuvieron una interrupción de la dosis de uno u otro fármaco debido a reacciones adversas. El cuarenta y dos por ciento de los pacientes del estudio tuvieron una reducción de la dosis de uno u otro fármaco debido a reacciones adversas. La tasa de interrupción debido a reacciones adversas fue del 11%.
En el ensayo 2, se evaluaron los datos de 450 pacientes (población de seguridad) que recibieron tratamiento con POMALYST + Dex de baja dosis (300 pacientes) o dexametasona de alta dosis (Dex de alta dosis) (150 pacientes). El número medio de ciclos de tratamiento para el brazo de POMALYST + Dex de baja dosis fue de 5. En el brazo de POMALYST + Dex de baja dosis, el 67% de los pacientes tuvieron una interrupción de la dosis de POMALYST, el tiempo medio hasta la primera interrupción de la dosis de POMALYST fue de 4,1 semanas. El veintisiete por ciento de los pacientes tuvieron una reducción de la dosis de POMALYST, el tiempo medio hasta la primera reducción de la dosis de POMALYST fue de 4,5 semanas. El ocho por ciento de los pacientes interrumpieron POMALYST debido a reacciones adversas.
Las tablas 3 y 4 resumen las reacciones adversas notificadas en los ensayos 1 y 2, respectivamente.
| * Independientemente de la atribución de la relación con POMALYST. a El brazo de POMALYST solo incluye a todos los pacientes aleatorizados al brazo de POMALYST solo que tomaron el fármaco del estudio; 61 de los 107 pacientes recibieron dexametasona durante el período de tratamiento. b Las reacciones adversas graves se notificaron en al menos 2 pacientes en cualquier brazo de tratamiento con POMALYST. Fecha límite de los datos: 01 de marzo de 2013 |
||||
|
Todas las reacciones adversas ≥10% en cualquiera de los brazos |
Grado 3 o 4 ≥5% en cualquiera de los brazos |
|||
|
Sistema corporal |
POMALYSTa |
POMALYST + Dex de baja dosis |
POMALYST |
POMALYST + Dex de baja dosis |
|
Número (%) de pacientes con al menos una reacción adversa |
107 (100) |
112 (100) |
98 (92) |
102 (91) |
|
Trastornos de la sangre y del sistema linfático |
||||
|
Neutropenia b |
57 (53) |
55 (49) |
51 (48) |
46 (41) |
|
Anemia b |
41 (38) |
47 (42) |
25 (23) |
24 (21) |
|
Trombocitopenia b |
28 (26) |
26 (23) |
24 (22) |
21 (19) |
|
Leucopenia |
14 (13) |
22 (20) |
7 (7) |
11 (10) |
|
Neutropenia febril b |
<10% |
<10% |
6 (6) |
3 (3) |
|
Linfopenia |
4 (4) |
17 (15) |
2 (2) |
8 (7) |
|
Trastornos generales y condiciones del lugar de administración |
||||
|
Fatiga y astenia b |
62 (58) |
70 (63) |
13 (12) |
19 (17) |
|
Edema periférico |
27 (25) |
19 (17) |
0 (0.0) |
0 (0.0) |
|
Pirexia b |
25 (23) |
36 (32) |
<5% |
<5% |
|
Escalofríos |
11 (10) |
14 (13) |
0 (0.0) |
0 (0.0) |
|
Trastornos gastrointestinales |
||||
|
Náuseas b |
39 (36) |
27 (24) |
<5% |
<5% |
|
Estreñimiento b |
38 (36) |
41 (37) |
<5% |
<5% |
|
Diarrea |
37 (35) |
40 (36) |
<5% |
<5% |
|
Vómitos b |
15 (14) |
16 (14) |
<5% |
0 (0.0) |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||||
|
Dolor de espalda b |
37 (35) |
36 (32) |
15 (14) |
11 (10) |
|
Dolor musculoesquelético en el pecho |
25 (23) |
22 (20) |
<5% |
0 (0.0) |
|
Espasmos musculares |
23 (21) |
22 (20) |
<5% |
<5% |
|
Artralgia |
18 (17) |
17 (15) |
<5% |
<5% |
|
Debilidad muscular |
15 (14) |
15 (13) |
6 (6) |
4 (4) |
|
Dolor óseo |
13 (12) |
8 (7) |
<5% |
<5% |
|
Dolor musculoesquelético |
13 (12) |
19 (17) |
<5% |
<5% |
|
Dolor en la extremidad |
8 (7) |
16 (14) |
0 (0.0) |
<5% |
|
Infecciones e infestaciones |
||||
|
Infección del tracto respiratorio superior |
40 (37) |
32 (29) |
<5% |
<5% |
|
Neumonía b |
30 (28) |
38 (34) |
21 (20) |
32 (29) |
|
Infección del tracto urinario b |
11 (10) |
19 (17) |
2 (2) |
10 (9) |
|
Sepsis b |
<10% |
<10% |
6 (6) |
5 (4) |
|
Trastornos del metabolismo y la nutrición |
||||
|
Disminución del apetito |
25 (23) |
21 (19) |
<5% |
0 (0.0) |
|
Hipercalcemia b |
23 (21) |
13 (12) |
11 (10) |
1 (<1) |
|
Hipokalemia |
13 (12) |
13 (12) |
<5% |
<5% |
|
Hiperglucemia |
12 (11) |
17 (15) |
<5% |
<5% |
|
Hiponatremia |
12 (11) |
14 (13) |
<5% |
<5% |
|
Deshidratación b |
<10% |
<10% |
5 (4.7) |
6 (5.4) |
|
Hipocalcemia |
6 (6) |
13 (12) |
0 (0.0) |
<5% |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||
|
Disnea b |
38 (36) |
50 (45) |
8 (7) |
14 (13) |
|
Tos |
18 (17) |
25 (22) |
0 (0.0) |
0 (0.0) |
|
Epistaxis |
18 (17) |
12 (11) |
<5% |
0 (0.0) |
|
Tos productiva |
10 (9) |
14 (13) |
0 (0.0) |
0 (0.0) |
|
Dolor orofaríngeo |
6 (6) |
12 (11) |
0 (0.0) |
0 (0.0) |
|
Trastornos del sistema nervioso |
||||
|
Vértigo |
24 (22) |
20 (18) |
<5% |
<5% |
|
Neuropatía periférica |
23 (21) |
20 (18) |
0 (0.0) |
0 (0.0) |
|
Cefalea |
16 (15) |
15 (13) |
0 (0.0) |
<5% |
|
Temblor |
11 (10) |
15 (13) |
0 (0.0) |
0 (0.0) |
|
Trastornos de la piel y del tejido subcutáneo |
||||
|
Erupción cutánea |
22 (21) |
18 (16) |
0 (0.0) |
<5% |
|
Prurito |
16 (15) |
10 (9) |
0 (0.0) |
0 (0.0) |
|
Piel seca |
10 (9) |
12 (11) |
0 (0.0) |
0 (0.0) |
|
Hiperhidrosis |
8 (7) |
18 (16) |
0 (0.0) |
0 (0.0) |
|
Sudoración nocturna |
5 (5) |
14 (13) |
0 (0.0) |
0 (0.0) |
|
Pruebas |
||||
|
Creatinina en sangre aumentada b |
20 (19) |
11 (10) |
6 (6) |
3 (3) |
|
Pérdida de peso |
16 (15) |
10 (9) |
0 (0.0) |
0 (0.0) |
|
Aumento de peso |
1 (<1) |
12 (11) |
0 (0.0) |
0 (0.0) |
|
Trastornos psiquiátricos |
||||
|
Ansiedad |
14 (13) |
8 (7) |
0 (0.0) |
0 (0.0) |
|
Estado confusional b |
13 (12) |
15 (13) |
6 (6) |
3 (3) |
|
Insomnio |
7 (7) |
18 (16) |
0 (0.0) |
0 (0.0) |
|
Trastornos renales y urinarios |
||||
|
Insuficiencia renal b |
16 (15) |
11 (10) |
9 (8) |
8 (7) |
| a El porcentaje no cumplió con los criterios para ser considerado como una reacción adversa para POMALYST para esa categoría de evento (es decir, todos los eventos adversos o eventos adversos de Grado 3 o 4). b Se informaron reacciones adversas graves en al menos 3 pacientes en el brazo POM + Dex de dosis baja, Y al menos 1% más que el porcentaje del brazo Dex de dosis alta. Corte de datos: 01 de marzo de 2013 |
||||
|
Todas las reacciones adversas |
Grado 3 o 4 |
|||
|
Sistema corporal |
POMALYST + Dex de dosis baja |
Dex de dosis alta |
POMALYST + Dex de dosis baja |
Dex de dosis alta |
|
Número (%) de pacientes con al menos una reacción adversa |
297 (99) |
149 (99) |
259 (86) |
127 (85) |
|
Trastornos de la sangre y del sistema linfático |
||||
|
Neutropenia b |
154 (51) |
31 (21) |
145 (48) |
24 (16) |
|
Trombocitopenia |
89 (30) a |
44 (29) a |
66 (22) a |
39 (26) a |
|
Leucopenia |
38 (13) |
8 (5) |
27 (9) |
5 (3) |
|
Neutropenia febril b |
28 (9) |
0 (0.0) |
28 (9) |
0 (0.0) |
|
Trastornos generales y condiciones del sitio de administración |
||||
|
Fatiga y astenia |
140 (47) |
64 (43) |
26 (9) a |
18 (12) a |
|
Pirexia b |
80 (27) |
35 (23) |
9 (3) a |
7 (5) a |
|
Edema periférico |
52 (17) |
17 (11) |
4 (1) a |
3 (2) a |
|
Dolor |
11 (4) a |
3 (2) a |
5 (2) |
1 (<1) |
|
Infecciones e infestaciones |
||||
|
Infección del tracto respiratorio superior b |
93 (31) |
19 (13) |
9 (3) |
1 (<1) |
|
Neumonía b |
58 (19) |
20 (13) |
47 (16) |
15 (10) |
|
Sepsis neutropénica b |
3 (1) a |
0 (0.0) a |
3 (1) |
0 (0.0) |
|
Trastornos gastrointestinales |
||||
|
Diarrea |
66 (22) |
28 (19) |
3 (1) a |
2 (1) a |
|
Estreñimiento |
65 (22) |
22 (15) |
7 (2) |
0 (0.0) |
|
Náuseas |
45 (15) |
17 (11) |
3 (1) a |
2 (1) a |
|
Vómitos |
23 (8) |
6 (4) |
3 (1) |
0 (0.0) |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||||
|
Dolor de espalda b |
59 (20) |
24 (16) |
15 (5) |
6 (4) |
|
Dolor óseo b |
54 (18) |
21 (14) |
22 (7) |
7 (5) |
|
Espasmos musculares |
46 (15) |
11 (7) |
1 (<1) a |
1 (<1) a |
|
Artralgia |
26 (9) |
7 (5) |
2 (<1) a |
1 (<1) a |
|
Dolor en la extremidad |
20 (7) a |
9 (6) a |
6 (2) |
0 (0.0) |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||
|
Disnea b |
76 (25) |
25 (17) |
17 (6) |
7 (5) |
|
Tos |
60 (20) |
15 (10) |
2 (<1) a |
1 (<1) a |
|
Enfermedad pulmonar obstructiva crónica b |
5 (2) a |
0 (0.0) a |
4 (1) |
0 (0.0) |
|
Trastornos del sistema nervioso |
||||
|
Neuropatía periférica |
52 (17) |
18 (12) |
5 (2) a |
2 (1) a |
|
Mareos |
37 (12) |
14 (9) |
4 (1) a |
2 (1) a |
|
Cefalea |
23 (8) |
8 (5) |
1 (<1) a |
0 (0.0) a |
|
Temblor |
17 (6) |
2 (1) |
2 (<1) a |
0 (0.0) a |
|
Depresión del nivel de conciencia |
5 (2) a |
0 (0.0) a |
3 (1) |
0 (0.0) |
|
Trastornos del metabolismo y la nutrición |
||||
|
Disminución del apetito |
38 (13) |
12 (8) |
3 (1) a |
2 (1) a |
|
Hipokalemia |
28 (9) a |
12 (8) a |
12 (4) |
4 (3) |
|
Hipocalcemia |
12 (4) a |
9 (6) a |
5 (2) |
1 (<1) |
|
Trastornos de la piel y del tejido subcutáneo |
||||
|
Erupción cutánea |
23 (8) |
2 (1) |
3 (1) |
0 (0.0) |
|
Prurito |
22 (7) |
5 (3) |
0 (0.0) a |
0 (0.0) a |
|
Hiperhidrosis |
15 (5) |
1 (<1) |
0 (0.0) a |
0 (0.0) a |
|
Pruebas de laboratorio |
||||
|
Conteo de neutrófilos disminuido |
15 (5) |
1 (<1) |
14 (5) |
1 (<1) |
|
Recuento de plaquetas disminuido |
10 (3) a |
3 (2) a |
8 (3) |
2 (1) |
|
Recuento de glóbulos blancos disminuido |
8 (3) a |
1 (<1) a |
8 (3) |
0 (0.0) |
|
Alanina aminotransferasa aumentada |
7 (2) a |
2 (1) a |
5 (2) |
0 (0.0) |
|
Aspartato aminotransferasa aumentada |
4 (1) a |
2 (1) a |
3 (1) |
0 (0.0) |
|
Recuento de linfocitos disminuido |
3 (1) a |
1 (<1) a |
3 (1) |
0 (0.0) |
|
Trastornos renales y urinarios |
||||
|
Insuficiencia renal |
31 (10) a |
18 (12) a |
19 (6) |
8 (5) |
|
Lesión, envenenamiento y complicaciones de procedimientos |
||||
|
Fractura de fémur b |
5 (2) a |
1 (<1) a |
5 (2) |
1 (<1) |
|
Trastornos del sistema reproductor femenino y de la mama |
||||
|
Dolor pélvico |
6 (2) a |
3 (2) a |
4 (1) |
0 (0.0) |
Otras reacciones adversas
Otras reacciones adversas de POMALYST en pacientes con MM, no descritas anteriormente, y consideradas importantes:
Trastornos cardíacos: Infarto de miocardio, Fibrilación auricular, Angina de pecho, Insuficiencia cardíaca congestiva
Trastornos del oído y el laberinto: Vértigo
Trastornos gastrointestinales: Dolor abdominal
Trastornos generales y condiciones del sitio de administración: Deterioro general de la salud física, Dolor torácico no cardíaco, Insuficiencia multiorgánica
Trastornos hepatobiliares: Hiperbilirrubinemia
Infecciones e infestaciones: Neumonía por Pneumocystis jiroveci, Infección por virus respiratorio sincitial, Sepsis neutropénica, Bacteriemia, Neumonía viral respiratoria sincitial, Celulitis, Urosepsis, Shock séptico, Colitis por Clostridium difficile, Neumonía estreptocócica, Neumonía lobar, Infección viral, Infección pulmonar
Investigaciones: Aumento de la alanina aminotransferasa, Disminución de la hemoglobina
Lesiones, envenenamiento y complicaciones de procedimientos: Caída, Fractura por compresión, Fractura por compresión vertebral
Trastornos del metabolismo y la nutrición: Hiperpotasemia, Falta de desarrollo
Trastornos del sistema nervioso: Depresión del nivel de conciencia, Síncope
Trastornos psiquiátricos: Cambio en el estado mental
Trastornos renales y urinarios: Retención urinaria, Hiponatremia
Trastornos del sistema reproductor y de las mamas: Dolor pélvico
Trastornos respiratorios, torácicos y mediastínicos: Enfermedad pulmonar intersticial, Embolia pulmonar, Insuficiencia respiratoria, Broncoespasmo
Trastornos vasculares: Hipotensión
Sarcoma de Kaposi (KS)
La seguridad de POMALYST en pacientes con KS se evaluó en el ensayo 12-C-0047 [ver Estudios clínicos (14.2)]. Veintiochos pacientes recibieron POMALYST 5 mg administrados por vía oral una vez al día los días 1 a 21 de ciclos repetidos de 28 días. El estudio excluyó a pacientes con trastornos procoagulantes o antecedentes de tromboembolismo venoso o arterial. Los pacientes recibieron profilaxis para la TVP con aspirina de dosis baja diaria. En todos los pacientes tratados en el ensayo 12-C-0047, el 75% estuvo expuesto a pomalidomida durante 6 meses o más y el 25% estuvo expuesto durante más de un año.
Las reacciones adversas graves ocurrieron en el 18% (5/28) de los pacientes que recibieron POMALYST. Las siguientes reacciones adversas graves ocurrieron en 1 paciente: anemia, disminución del recuento de neutrófilos y hematuria.
La interrupción permanente debido a una reacción adversa ocurrió en el 11% (3/28) de los pacientes que recibieron POMALYST.
Las interrupciones de la dosis debido a una reacción adversa ocurrieron en el 14% (4/28) de los pacientes que recibieron POMALYST. La reacción adversa más frecuente que requirió interrupción de la dosis fue la disminución del recuento de neutrófilos, que ocurrió en 3 pacientes.
La dosis de POMALYST se redujo debido a una reacción adversa en 1 paciente debido a la gota.
Las tablas 5 y 6 resumen las reacciones adversas y las anomalías de laboratorio selectas notificadas en el ensayo 12-C-0047.
|
Reacción adversa |
Grados 1-4 |
Grado 3 o 4 |
|
Erupción, maculo-papular |
71 |
3.6 |
|
Estreñimiento |
71 |
0 |
|
Fatiga |
68 |
0 |
|
Náuseas |
36 |
0 |
|
Diarrea |
32 |
3.6 |
|
Tos |
29 |
0 |
|
Disnea |
29 |
0 |
|
Edema periférico |
29 |
3.6 |
|
Infección de las vías respiratorias altas |
29 |
0 |
|
Espasmos musculares |
25 |
0 |
|
Hipotiroidismo |
21 |
0 |
|
Piel seca |
21 |
0 |
|
Escalofríos |
21 |
0 |
| * El denominador es el número de pacientes para los que hay una evaluación inicial y al menos una evaluación posterior a la inicial para el parámetro de laboratorio. | ||
|
Anomalía de laboratorio |
Grados 1-4* |
Grados 3-4* |
|
Hematología |
||
|
Conteo de neutrófilos absolutos disminuido |
96 |
50 |
|
Glóbulos blancos disminuidos |
79 |
3.6 |
|
Hemoglobina disminuida |
54 |
0 |
|
Plaquetas disminuidas |
54 |
0 |
|
Química |
||
|
Creatinina elevada |
86 |
3.6 |
|
Glucosa elevada |
57 |
7 |
|
Albúmina disminuida |
54 |
0 |
|
Fósforo disminuido |
54 |
25 |
|
Calcio disminuido |
50 |
0 |
|
Alanina aminotransferasa (ALT) aumentada |
32 |
0 |
|
Aspartato aminotransferasa (AST) aumentada |
25 |
0 |
|
Creatina quinasa elevada |
25 |
7 |
|
Magnesio disminuido |
14 |
0 |
|
Fosfatasa alcalina elevada |
14 |
3.6 |
6.2 Experiencia Postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso post-aprobación de POMALYST. Debido a que estas reacciones se reportan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Trastornos de la sangre y del sistema linfático: Pancitopenia
Trastornos endocrinos: Hipotiroidismo, hipertiroidismo
Trastornos gastrointestinales: Hemorragia gastrointestinal
Trastornos hepatobiliares: Insuficiencia hepática (incluidos casos fatales), elevación de las enzimas hepáticas
Trastornos del sistema inmunitario: Reacciones alérgicas (por ejemplo, angioedema, anafilaxia, urticaria), rechazo de trasplante de órgano sólido
Infecciones e infestaciones: Reactivación del virus de la hepatitis B, Herpes zoster, leucoencefalopatía multifocal progresiva (PML)
Neoplasias benignas, malignas y no especificadas (incluidos quistes y pólipos): Síndrome de lisis tumoral, carcinoma de células basales y carcinoma de células escamosas de la piel
Trastornos de la piel y del tejido subcutáneo: Síndrome de Stevens-Johnson, necrólisis epidérmica tóxica, reacción medicamentosa con eosinofilia y síntomas sistémicos (DRESS)
7 INTERACCIONES MEDICAMENTOSAS
7.1 Medicamentos Que Afectan Las Concentraciones Plasmáticas De Pomalidomida
Inhibidores de CYP1A2:
En sujetos sanos, la administración conjunta de fluvoxamina, un potente inhibidor de CYP1A2, aumentó la Cmáx y el AUC de pomalidomida en un 24 % y un 125 % respectivamente [ver Farmacología Clínica (12.3)]. El aumento de la exposición a pomalidomida puede aumentar el riesgo de toxicidades relacionadas con la exposición. Evite la administración conjunta de inhibidores potentes de CYP1A2 (p. ej., ciprofloxacino y fluvoxamina). Si no se puede evitar la administración conjunta, reduzca la dosis de POMALYST [ver Dosificación y administración (2.6)].
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Registro de exposición al embarazo
Existe un registro de exposición al embarazo que monitorea los resultados del embarazo en mujeres expuestas a POMALYST durante el embarazo, así como en las parejas femeninas de pacientes masculinos que están expuestos a POMALYST. Este registro también se utiliza para comprender la causa raíz del embarazo. Informe cualquier sospecha de exposición fetal a POMALYST a la FDA a través del programa MedWatch al 1-800-FDA-1088 y también al Centro de llamadas de REMS al 1-888-423-5436.
Resumen de riesgos
Con base en el mecanismo de acción [ver Farmacología clínica (12.1)] y los hallazgos de estudios en animales, POMALYST puede causar daño embrionario-fetal cuando se administra a una mujer embarazada y está contraindicado durante el embarazo [ver Contraindicaciones (4), y Advertencias y precauciones (5.1)].
POMALYST es un análogo de la talidomida. La talidomida es un teratógeno humano, que induce una alta frecuencia de defectos de nacimiento graves y potencialmente mortales, como amelia (ausencia de extremidades), focomelia (extremidades cortas), hipoplasia de los huesos, ausencia de huesos, anomalías del oído externo (incluida la anotia, micropinna, conductos auditivos externos pequeños o ausentes), parálisis facial, anomalías oculares (anoftalmos, microftalmos) y defectos cardíacos congénitos. También se han documentado malformaciones del tracto digestivo, del tracto urinario y genitales, y se ha informado mortalidad en o poco después del nacimiento en aproximadamente el 40% de los bebés.
La pomalidomida fue teratógena tanto en ratas como en conejos cuando se administró durante el período de organogénesis. La pomalidomida cruzó la placenta después de la administración a conejos preñados (ver Datos). Si este medicamento se usa durante el embarazo o si la paciente queda embarazada mientras toma este medicamento, se le debe informar del riesgo potencial para el feto.
Si se produce un embarazo durante el tratamiento, suspenda inmediatamente el medicamento. En estas condiciones, refiera a la paciente a un obstetra/ginecólogo con experiencia en toxicidad reproductiva para una mayor evaluación y asesoramiento. Informe cualquier sospecha de exposición fetal a POMALYST a la FDA a través del programa MedWatch al 1-800-FDA-1088 y también al Centro de llamadas de REMS al 1-888-423-5436.
El riesgo de fondo estimado de defectos de nacimiento mayores y aborto espontáneo para la población indicada es desconocido. El riesgo de fondo estimado en la población general de EE. UU. de defectos de nacimiento mayores es del 2% al 4% y de aborto espontáneo del 15% al 20% de los embarazos clínicamente reconocidos.
Datos
Datos de animales
La pomalidomida fue teratógena tanto en ratas como en conejos en los estudios de desarrollo embrionario-fetal cuando se administró durante el período de organogénesis.
En ratas, la pomalidomida se administró por vía oral a animales preñados a dosis de 25 a 1000 mg/kg/día. Se observaron malformaciones o ausencia de vejiga urinaria, ausencia de glándula tiroides y fusión y desalineación de los elementos vertebrales lumbares y torácicos (arcos vertebrales, centrales y/o neurales) en todos los niveles de dosis. No se observó toxicidad materna en este estudio. La dosis más baja en ratas resultó en una exposición (AUC) aproximadamente 85 veces mayor que la exposición humana a la dosis recomendada de 4 mg/día. Otras toxicidades embrionarias-fetales incluyeron un aumento de las resorciones que llevaron a una disminución del número de fetos viables.
En conejos, la pomalidomida se administró por vía oral a animales preñados a dosis de 10 a 250 mg/kg/día. Se observó un aumento de las malformaciones cardíacas, como el defecto del tabique interventricular, en todas las dosis, con aumentos significativos a 250 mg/kg/día. Las malformaciones adicionales observadas a 250 mg/kg/día incluyeron anomalías en las extremidades (extremidades anteriores y/o posteriores flexionadas y/o rotadas, dígitos no unidos o ausentes) y malformaciones esqueléticas asociadas (metacarpo no osificado, falange y metacarpo desalineado, dígito ausente, falange no osificada y tibia corta no osificada o doblada), dilatación moderada del ventrículo lateral en el cerebro, colocación anormal de la arteria subclavia derecha, lóbulo intermedio ausente en los pulmones, riñón bajo, morfología hepática alterada, pelvis incompletamente o no osificada, un promedio aumentado de costillas torácicas supernumerarias y un promedio reducido de tarsos osificados. No se observó toxicidad materna a la dosis baja (10 mg/kg/día) que resultó en anomalías cardíacas en los fetos; esta dosis resultó en una exposición (AUC) aproximadamente igual a la reportada en humanos a la dosis recomendada de 4 mg/día. La toxicidad embrionaria-fetal adicional incluyó un aumento de la resorción.
Después de la administración oral diaria de pomalidomida desde el día 7 de gestación hasta el día 20 de gestación en conejos preñados, las concentraciones plasmáticas fetales de pomalidomida fueron aproximadamente el 50% de la Cmax materna a todas las dosis (5 a 250 mg/kg/día), lo que indica que la pomalidomida cruzó la placenta.
8.2 Lactancia
Resumen de riesgos
No hay información sobre la presencia de pomalidomida en la leche materna, los efectos de POMALYST en el niño amamantado o los efectos de POMALYST en la producción de leche. La pomalidomida se excretó en la leche de ratas lactantes (ver Datos). Debido a que muchos medicamentos se excretan en la leche materna y debido al potencial de reacciones adversas en un niño amamantado por POMALYST, se debe aconsejar a las mujeres que no amamanten durante el tratamiento con POMALYST.
Datos
Datos de animales
Después de una sola administración oral de pomalidomida a ratas lactantes aproximadamente 14 días después del parto, la pomalidomida se transfirió a la leche, con relaciones leche-plasma de 0,63 a 1,46.
8.3 Mujeres y hombres en edad fértil
Prueba de embarazo
POMALYST puede causar daño fetal cuando se administra durante el embarazo [ver Uso en poblaciones específicas (8.1)]. Verifique el estado de embarazo de las mujeres en edad fértil antes de iniciar el tratamiento con POMALYST y durante el tratamiento. Avise a las mujeres en edad fértil que deben evitar el embarazo 4 semanas antes del tratamiento, mientras toman POMALYST, durante las interrupciones de la dosis y durante al menos 4 semanas después de completar el tratamiento.
Las mujeres en edad fértil deben tener 2 pruebas de embarazo negativas antes de iniciar POMALYST. La primera prueba debe realizarse dentro de los 10-14 días, y la segunda prueba dentro de las 24 horas previas a la prescripción de POMALYST. Una vez que el tratamiento ha comenzado y durante las interrupciones de la dosis, las pruebas de embarazo para las mujeres en edad fértil deben realizarse semanalmente durante las primeras 4 semanas de uso, luego las pruebas de embarazo deben repetirse cada 4 semanas en las mujeres con ciclos menstruales regulares. Si los ciclos menstruales son irregulares, las pruebas de embarazo deben realizarse cada 2 semanas. Las pruebas de embarazo y el asesoramiento deben realizarse si una paciente pierde su período o si hay alguna anormalidad en su sangrado menstrual. El tratamiento con POMALYST debe interrumpirse durante esta evaluación.
Anticoncepción
Mujeres
Las mujeres en edad fértil deben comprometerse a abstenerse continuamente de las relaciones sexuales heterosexuales o a utilizar 2 métodos de control de la natalidad fiables simultáneamente: una forma de anticoncepción altamente eficaz – ligadura de trompas, DIU, hormonal (píldoras anticonceptivas, inyecciones, parches hormonales, anillos vaginales o implantes), o vasectomía de la pareja, y 1 método anticonceptivo eficaz adicional – condón masculino de látex o sintético, diafragma o capuchón cervical. La anticoncepción debe comenzar 4 semanas antes de iniciar el tratamiento con POMALYST, durante el tratamiento, durante las interrupciones de la dosis y continuar durante 4 semanas después de la interrupción del tratamiento con POMALYST. La anticoncepción fiable está indicada incluso cuando ha habido antecedentes de infertilidad, a menos que se deba a una histerectomía. Las mujeres en edad fértil deben ser derivadas a un proveedor cualificado de métodos anticonceptivos, si es necesario.
Hombres
La pomalidomida está presente en el semen de los hombres que toman POMALYST. Por lo tanto, los hombres deben usar siempre un condón de látex o sintético durante cualquier contacto sexual con mujeres en edad fértil mientras toman POMALYST y hasta 4 semanas después de interrumpir POMALYST, incluso si se han sometido a una vasectomía exitosa. Los pacientes masculinos que toman POMALYST no deben donar esperma.
Infertilidad
Basándose en los hallazgos en animales, la fertilidad femenina puede verse comprometida por el tratamiento con POMALYST [ver Toxicología no clínica (13.1)].
8.4 Uso pediátrico
La seguridad y eficacia de POMALYST no se han establecido en pacientes pediátricos. La seguridad y eficacia se evaluaron pero no se establecieron en dos estudios abiertos: un estudio de escalada de dosis en 25 pacientes pediátricos de 5 a <17 años con tumores del SNC recurrentes, progresivos o refractarios [NCT02415153] y un estudio de grupos paralelos realizado en 47 pacientes pediátricos de 4 a <17 años con glioma de alto grado recurrente o progresivo, meduloblastoma, ependimoma o glioma pontino intrínseco difuso (DIPG) [NCT03257631]. No se observaron nuevas señales de seguridad en los pacientes pediátricos en estos estudios.
A la misma dosis por superficie corporal, la exposición a la pomalidomida en 55 pacientes pediátricos de 4 a < 17 años de edad estuvo dentro del rango observado en pacientes adultos con MM pero superior a la exposición observada en pacientes adultos con KS [ver Farmacología clínica (12.3)].
8.5 Uso geriátrico
Mieloma múltiple
Del total de pacientes en los estudios clínicos de POMALYST, el 44% tenía más de 65 años, mientras que el 10% tenía más de 75 años. No se observaron diferencias generales en la eficacia entre estos pacientes y los pacientes más jóvenes. En estos estudios, los pacientes mayores de 65 años tenían más probabilidades que los pacientes menores o iguales a 65 años de experimentar neumonía.
8.6 Insuficiencia renal
En pacientes con insuficiencia renal grave que requieren diálisis, el AUC de pomalidomida aumentó un 38% y la tasa de SAE aumentó un 64% en relación con los pacientes con función renal normal; por lo tanto, se recomienda el ajuste de la dosis inicial. Para los pacientes con insuficiencia renal grave que requieren diálisis, administre POMALYST después de completar la hemodiálisis en los días de diálisis porque la exposición a la pomalidomida podría disminuir significativamente durante la diálisis [ver Dosificación y administración (2.7) y Farmacología clínica (12.3)].
8.7 Insuficiencia hepática
Pomalidomida se metaboliza principalmente en el hígado. Después de la administración de una dosis única, el AUC de pomalidomida aumentó un 51%, un 58% y un 72% en sujetos con insuficiencia hepática leve (clase A de Child-Pugh), moderada (clase B de Child-Pugh) y grave (clase C de Child-Pugh), respectivamente, en comparación con los sujetos con función hepática normal. Se recomienda el ajuste de la dosis en pacientes con insuficiencia hepática [ver Dosificación y administración (2.8) y Farmacología clínica (12.3)].
8.8 Fumar tabaco
Fumar cigarrillos reduce el AUC de pomalidomida debido a la inducción del CYP1A2. Avise a los pacientes que fumar puede reducir la eficacia de pomalidomida [ver Farmacología clínica (12.3)].
10 SOBREDOSIS
La hemodiálisis puede eliminar la pomalidomida de la circulación.
11 DESCRIPCIÓN
La pomalidomida es un análogo de la talidomida. El nombre químico es (RS)-4-Amino-2-(2,6-dioxo-piperidin-3-il)-isoindolina-1,3-diona y tiene la siguiente estructura química:
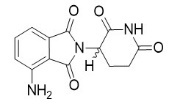
La fórmula empírica de la pomalidomida es C13H11N3O4 y el peso molecular en gramos es 273,24.
La pomalidomida es un polvo sólido amarillo. Tiene una solubilidad limitada a baja en solventes orgánicos y una baja solubilidad en todas las soluciones de pH (aproximadamente 0,01 mg/mL). La pomalidomida tiene un átomo de carbono quiral que existe como una mezcla racémica de los enantiómeros R(+) y S(-).
POMALYST está disponible en cápsulas de 1 mg, 2 mg, 3 mg y 4 mg para administración oral. Cada cápsula contiene pomalidomida como ingrediente activo y los siguientes ingredientes inactivos: manitol, almidón pregelatinizado y fumarato de estearilo sódico. La cápsula de 1 mg contiene gelatina, dióxido de titanio, FD&C azul 2, óxido de hierro amarillo, tinta blanca e tinta negra. La cápsula de 2 mg contiene gelatina, dióxido de titanio, FD&C azul 2, óxido de hierro amarillo, FD&C rojo 3 y tinta blanca. La cápsula de 3 mg contiene gelatina, dióxido de titanio, FD&C azul 2, óxido de hierro amarillo y tinta blanca. La cápsula de 4 mg contiene gelatina, dióxido de titanio, FD&C azul 1, FD&C azul 2 y tinta blanca.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Pomalidomida es un análogo de la talidomida con propiedades inmunomoduladoras, antiangiogénicas y antineoplásicas. Las actividades celulares de la pomalidomida están mediadas a través de su diana cereblon, un componente de un complejo enzimático de ligasa de ubiquitina E3 de anillo de cullin. In vitro, en presencia del fármaco, las proteínas sustrato (incluidas Aiolos e Ikaros) se dirigen a la ubiquitinación y posterior degradación, lo que lleva a efectos citotóxicos e inmunomoduladores directos. En ensayos celulares in vitro, la pomalidomida inhibió la proliferación e indujo la apoptosis de las células tumorales hematopoyéticas. Además, la pomalidomida inhibió la proliferación de líneas celulares de mieloma múltiple (MM) resistentes a lenalidomida y sinergizó con dexametasona tanto en líneas celulares sensibles a lenalidomida como resistentes a lenalidomida para inducir la apoptosis de las células tumorales. La pomalidomida mejoró la inmunidad mediada por células T y células asesinas naturales (NK) e inhibió la producción de citocinas proinflamatorias (por ejemplo, TNF-α e IL-6) por los monocitos. La pomalidomida demostró actividad antiangiogénica en un modelo de tumor de ratón y en el modelo in vitro de cordón umbilical.
12.2 Farmacodinamia
Los análisis de exposición-respuesta a la pomalidomida mostraron que no había relación entre el nivel de exposición sistémica a la pomalidomida y la eficacia o seguridad después de una dosis de pomalidomida de 4 mg.
Electrofisiología cardíaca
El potencial de prolongación del QTc de la pomalidomida se evaluó en un estudio cruzado, aleatorizado, doble ciego de un solo centro (N=72) utilizando 4 mg de pomalidomida, 20 mg de pomalidomida, placebo y 400 mg de moxifloxacina (control positivo). No se observó ningún efecto significativo de prolongación del QTc de la pomalidomida después de dosis de pomalidomida de 4 y 20 mg.
12.3 Farmacocinética
En pacientes con MM que recibieron POMALYST 4 mg diarios solos o en combinación con dexametasona, la exposición al fármaco en estado estacionario de la pomalidomida se caracterizó por un AUC (CV%) de 860 (37%) ng∙h/mL y una Cmax (CV%) de 75 (32%) ng/mL. En pacientes con sarcoma de Kaposi (KS) que recibieron POMALYST 5 mg diarios, la exposición al fármaco en estado estacionario de la pomalidomida se caracterizó por un AUC de 462,3 ng∙h/mL (82%) y una Cmax de 53,1 ng/mL (50%).
Absorción
Después de la administración de dosis orales únicas de POMALYST, la concentración plasmática máxima (Cmax) de la pomalidomida se produce entre 2 y 3 horas después de la dosis en pacientes con MM o KS.
Efecto de los alimentos
La coadministración de POMALYST con una comida rica en grasas (aproximadamente el 50% del contenido calórico total) y una comida rica en calorías (aproximadamente 800 a 1000 calorías) (la comida contenía aproximadamente 150, 250 y 500 a 600 calorías de proteínas, carbohidratos y grasas, respectivamente) retrasa el Tmax en 2,5 horas, disminuyó la Cmax plasmática media y el AUC en sujetos sanos en aproximadamente un 27% y un 8%, respectivamente.
Distribución
La pomalidomida tiene un volumen de distribución aparente medio (Vd/F) entre 62 y 138 L en estado estacionario en pacientes con MM o KS.
La pomalidomida se distribuye en el semen de sujetos sanos a una concentración de aproximadamente el 67% del nivel plasmático a las 4 horas después de la dosis (~Tmax) después de 4 días de dosificación de 2 mg una vez al día.
La unión a proteínas plasmáticas humanas de la pomalidomida oscila entre el 12% y el 44% y no depende de la concentración. La pomalidomida es un sustrato de la P-gp.
Eliminación
La pomalidomida tiene un aclaramiento corporal total medio (CL/F) de 7-10 L/h en pacientes con MM o KS. La pomalidomida se elimina con una vida media plasmática mediana de 9,5 horas en sujetos sanos y 7,5 horas en pacientes con MM o KS.
Metabolismo
La pomalidomida se metaboliza principalmente en el hígado por CYP1A2 y CYP3A4. También se observaron contribuciones menores de CYP2C19 y CYP2D6 in vitro.
Excreción
Después de una administración oral única de [14C]-pomalidomida a sujetos sanos, aproximadamente el 73% y el 15% de la dosis radiactiva se eliminó en la orina y las heces, respectivamente, con aproximadamente el 2% y el 8% de la dosis radiactiva eliminada sin cambios como pomalidomida en la orina y las heces.
Poblaciones específicas
La edad (de 61 a 85 años), el sexo y la raza no tienen un efecto clínicamente significativo en la exposición sistémica a la pomalidomida.
Pacientes con insuficiencia renal
Los parámetros farmacocinéticos de la pomalidomida no se vieron afectados significativamente en pacientes con insuficiencia renal moderada (30 mL/min ≤ CLcr< 60 mL/min) o grave (15 mL/min ≤ CLcr< 30 mL/min) en relación con los pacientes con función renal normal (CLcr ≥ 60 mL/min). La exposición media (AUC) a la pomalidomida aumentó un 38% en pacientes con insuficiencia renal grave que requerían diálisis (CLcr< 30 mL/min que requieren diálisis) y un 40% en pacientes con enfermedad renal en estadio terminal (CLcr< 15 mL/min) en días sin diálisis. En pacientes con insuficiencia renal grave que requieren diálisis, el aclaramiento dialítico estimado es de aproximadamente 12 L/h, que es superior al aclaramiento corporal total de la pomalidomida, lo que indica que la hemodiálisis eliminará la pomalidomida de la circulación sanguínea.
Estudios de interacción medicamentosa
Estudios clínicos
La coadministración de POMALYST con los siguientes fármacos no aumentó la exposición a pomalidomida hasta un grado clínicamente significativo: ketoconazol (un inhibidor potente de CYP3A4 y P-gp), carbamazepina (un inductor potente de CYP3A4) y dexametasona (un inductor débil a moderado de CYP3A4). No se ha estudiado la coadministración de POMALYST con fármacos que son inductores de CYP1A2.
Inhibidores de CYP1A2: La coadministración de fluvoxamina (un inhibidor potente de CYP1A2) con POMALYST aumentó la exposición media [intervalo de confianza del 90%] a pomalidomida en un 125% [98% a 157%] en comparación con POMALYST solo en sujetos sanos. La coadministración de fluvoxamina en presencia de ketoconazol (un inhibidor potente de CYP3A4 y P-gp) con POMALYST aumentó la exposición media a pomalidomida en un 146% [126% a 167%] en comparación con POMALYST administrado solo en sujetos sanos, lo que indica el efecto predominante de la inhibición de CYP1A2 en el aumento de la exposición a pomalidomida [ver Dosificación y administración (2.6) y Interacciones medicamentosas (7.1)].
Inhibidores potentes de CYP3A4 y P-gp: La coadministración de ketoconazol (un inhibidor potente de CYP3A4 y P-gp) en 16 sujetos masculinos sanos aumentó el AUC de pomalidomida en un 19% en comparación con POMALYST administrado solo.
Inductores potentes de CYP1A2: No se ha estudiado la coadministración de POMALYST con fármacos que son inductores de CYP1A2 y puede reducir la exposición a pomalidomida.
Inductores potentes de CYP3A4: La coadministración de carbamazepina a 16 sujetos masculinos sanos disminuyó el AUC de pomalidomida en un 20% con un intervalo de confianza del 90% [13% a 27%] en comparación con cuando se administró pomalidomida solo.
Dexametasona: La coadministración de dosis múltiples de 4 mg de POMALYST con 20 mg a 40 mg de dexametasona (un inductor débil a moderado de CYP3A4) a pacientes con MM no tuvo ningún efecto en la farmacocinética de pomalidomida en comparación con cuando se administró pomalidomida solo.
Fumar: En 14 sujetos masculinos sanos que fumaron 25 cigarrillos al día durante un total de 10 días, después de una dosis oral única de 4 mg de POMALYST, la Cmax de pomalidomida aumentó un 14% mientras que el AUC de pomalidomida disminuyó un 32%, en comparación con la de 13 sujetos masculinos sanos que no fumaban.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No se han realizado estudios que examinen el potencial carcinogénico de la pomalidomida. Uno de los 12 monos que recibieron una dosis de 1 mg/kg de pomalidomida (una exposición aproximadamente 15 veces mayor que la exposición en pacientes con la dosis recomendada de 4 mg/día) desarrolló leucemia mieloide aguda en un estudio de toxicología de dosis repetidas de 9 meses.
La pomalidomida no fue mutagénica ni clastogénica en una batería de pruebas, incluido el ensayo de mutación inversa en bacterias (prueba de Ames), el ensayo in vitro con linfocitos de sangre periférica humana y la prueba de micronúcleos en ratas tratadas por vía oral con dosis de hasta 2000 mg/kg/día.
En un estudio de fertilidad y desarrollo embrionario temprano en ratas, los machos tratados con el fármaco se aparearon con hembras tratadas o no tratadas. Se administró pomalidomida a machos y hembras en dosis de 25 a 1000 mg/kg/día. Cuando los machos tratados se aparearon con hembras tratadas, hubo un aumento en la pérdida posterior a la implantación y una disminución en el número medio de embriones viables en todos los niveles de dosis. No hubo otros efectos sobre las funciones reproductivas o el número de embarazos. La dosis más baja probada en animales resultó en una exposición (AUC) aproximadamente 100 veces mayor que la exposición en pacientes con la dosis recomendada de 4 mg/día. Cuando los machos tratados en este estudio se aparearon con hembras no tratadas, todos los parámetros uterinos fueron comparables a los de los controles. Con base en estos resultados, los efectos observados se atribuyeron al tratamiento de las hembras.
14 ESTUDIOS CLÍNICOS
14.1 Mieloma múltiple
Estudio 1
El estudio 1 fue un estudio abierto, aleatorizado, multicéntrico, de fase 2 en pacientes con mieloma múltiple (MM) recidivante que eran refractarios a su último tratamiento para el mieloma y habían recibido lenalidomida y bortezomib. Se consideró que los pacientes tenían una recidiva si habían alcanzado al menos una enfermedad estable durante al menos 1 ciclo de tratamiento para al menos 1 régimen anterior y luego desarrollaron una enfermedad progresiva. Se consideró que los pacientes eran refractarios si experimentaban progresión de la enfermedad durante o dentro de los 60 días posteriores a su último tratamiento. Un total de 221 pacientes fueron asignados aleatoriamente para recibir POMALYST solo o POMALYST con dosis bajas de Dex. En el estudio 1, se evaluó la seguridad y la eficacia de POMALYST 4 mg, una vez al día durante 21 de 28 días, hasta la progresión de la enfermedad, solo y en combinación con dosis bajas de Dex (40 mg/día administrados solo los días 1, 8, 15 y 22 de cada ciclo de 28 días para pacientes de 75 años o menos, o 20 mg/día administrados solo los días 1, 8, 15 y 22 de cada ciclo de 28 días para pacientes mayores de 75 años). A los pacientes del grupo de POMALYST solo se les permitió agregar dosis bajas de Dex al progresar la enfermedad.
La Tabla 7 resume las características basales del paciente y de la enfermedad en el Estudio 1. Las características demográficas y de la enfermedad al inicio del estudio fueron equilibradas y comparables entre los grupos de estudio.
| POMALYST (n=108) |
POMALYST + dosis bajas de Dex (n=113) |
|
|---|---|---|
| Fecha de corte de datos: 01 de abril de 2011 | ||
|
Características del paciente |
||
|
Mediana de edad, años (rango) |
61 (37-88) |
64 (34-88) |
|
Distribución por edad, n (%) |
||
|
<65 años |
65 (60.2) |
60 (53.1) |
|
≥65 años |
43 (39.8) |
53 (46.9) |
|
Sexo, n (%) |
||
|
Masculino |
57 (52.8) |
62 (54.9) |
|
Femenino |
51 (47.2) |
51 (45.1) |
|
Raza/etnia, n (%) |
||
|
Blanca |
86 (79.6) |
92 (81.4) |
|
Negra o afroamericana |
16 (14.8) |
17 (15) |
|
Todas las demás razas |
6 (5.6) |
4 (3.6) |
|
ECOG Performance, n (%) |
||
|
Estado 0-1 |
95 (87.9) |
100 (88.5) |
|
Características de la enfermedad |
||
|
Número de terapias previas |
||
|
Mediana (mín., máx.) |
5 (2, 12) |
5 (2, 13) |
|
Trasplante previo, n (%) |
82 (75.9) |
84 (74.3) |
|
Refractario a bortezomib y lenalidomida, n (%) |
64 (59.3) |
69 (61.1) |
La Tabla 8 resume los resultados del análisis de la tasa de respuesta global (TRG) y la duración de la respuesta (DR), según las evaluaciones del Comité de Adjudicación de Revisión Independiente para los grupos de tratamiento en el Ensayo 1. La TRG no difirió según el tipo de tratamiento anti mieloma previo.
| a Resultados previos a la adición de dexametasona. b TRG = RP + RC según los criterios del EBMT. IC, intervalo de confianza; NE, no establecido (aún no se ha alcanzado la mediana). Fecha de corte de datos: 01 de abril de 2011 |
||
|
POMALYSTa |
POMALYST + Dosis baja de Dex |
|
|
Respuesta |
||
|
Tasa de respuesta global (TRG),b n (%) |
8 (7.4) |
33 (29.2) |
|
IC del 95% para la TRG (%) |
(3.3, 14.1) |
(21.0, 38.5) |
|
Respuesta completa (RC), n (%) |
0 (0.0) |
1 (0.9) |
|
Respuesta parcial (RP), n (%) |
8 (7.4) |
32 (28.3) |
|
Duración de la respuesta (DR) |
||
|
Mediana, meses |
NE |
7.4 |
|
IC del 95% para la DR (meses) |
NE |
(5.1, 9.2) |
Ensayo 2
El ensayo 2 fue un estudio de fase 3, multicéntrico, aleatorizado, abierto, en el que se comparó el tratamiento con POMALYST + dosis baja de Dex con dosis alta de Dex en pacientes adultos con MM recidivante y refractario, que habían recibido al menos dos regímenes de tratamiento previos, incluidos lenalidomida y bortezomib, y que habían presentado progresión de la enfermedad en los 60 días posteriores a la última terapia o antes. Los pacientes con un aclaramiento de creatinina ≥ 45 ml/min cumplían los requisitos para participar en el ensayo. En el ensayo se incluyó a un total de 455 pacientes: 302 en el grupo de POMALYST + dosis baja de Dex y 153 en el grupo de dosis alta de Dex. Los pacientes del grupo de POMALYST + dosis baja de Dex recibieron 4 mg de POMALYST por vía oral los días 1 a 21 de cada ciclo de 28 días. La dexametasona (40 mg) se administró una vez al día los días 1, 8, 15 y 22 de un ciclo de 28 días. Los pacientes > 75 años comenzaron el tratamiento con 20 mg de dexametasona con la misma pauta. En el grupo de dosis alta de Dex, la dexametasona (40 mg) se administró una vez al día los días 1 a 4, 9 a 12 y 17 a 20 de un ciclo de 28 días. Los pacientes > 75 años comenzaron el tratamiento con 20 mg de dexametasona con la misma pauta. El tratamiento continuó hasta que los pacientes presentaron progresión de la enfermedad.
Las características basales de los pacientes y de la enfermedad estaban equilibradas y fueron comparables entre los grupos del estudio, como se resume en la Tabla 9. En general, el 94 % de los pacientes presentaban una enfermedad refractaria a lenalidomida, el 79 % presentaban una enfermedad refractaria a bortezomib y el 74 % presentaban una enfermedad refractaria tanto a lenalidomida como a bortezomib.
| Fecha de corte de datos: 01 de marzo de 2013 | ||
|
POMALYST + Dosis baja de Dex |
Dosis alta de Dex |
|
|
(N=302) |
(N=153) |
|
|
Características del paciente |
||
|
Mediana de edad, años (intervalo) |
64 (35, 84) |
65 (35, 87) |
|
Distribución por edad n (%) |
||
|
< 65 años |
158 (52) |
74 (48) |
|
≥ 65 años |
144 (48) |
79 (52) |
|
Sexo n (%) |
||
|
Hombre |
181 (60) |
87 (57) |
|
Mujer |
121 (40) |
66 (43) |
|
Raza/etnia n (%) |
||
|
Blanca |
244 (81) |
113 (74) |
|
Negra o afroamericana |
4 (1) |
3 (2) |
|
Asiática |
4 (1) |
0 (0) |
|
Otra raza |
2 (1) |
2 (1) |
|
No recogida |
48 (16) |
35 (23) |
|
ECOG Performance n (%) |
||
|
Estado 0 |
110 (36) |
36 (24) |
|
Estado 1 |
138 (46) |
86 (56) |
|
Estado 2 |
52 (17) |
25 (16) |
|
Estado 3 |
0 (0) |
3 (2) |
|
Faltante |
2 (1) |
3 (2) |
|
Características de la enfermedad |
||
|
Número de terapias previas |
||
|
Media, (mín., máx.) |
5 (2, 14) |
5 (2, 17) |
|
Trasplante de células madre previo n (%) |
214 (71) |
105 (69) |
|
Refractario a bortezomib y lenalidomida n (%) |
225 (75) |
113 (74) |
La Tabla 10 resume la supervivencia libre de progresión (SLP) y la tasa de respuesta global (TRG) según la evaluación de la revisión del Comité de Adjudicación de Revisión Independiente (CARI) en el análisis final de SLP y la supervivencia global (SG) en el análisis de SG. La SLP fue significativamente más prolongada con POMALYST + dosis baja de Dex que con dosis alta de Dex: HR 0.45 (IC del 95%: 0.35-0.59 p < 0.001). La SG también fue significativamente más prolongada con POMALYST + dosis baja de Dex que con dosis alta de Dex: HR 0.70 (IC del 95%: 0.54-0.92 p = 0.009). Las curvas de Kaplan-Meier para la SLP y la SG para la población por intención de tratar (ITT) se proporcionan en las Figuras 1 y 2, respectivamente.
| Nota: IC = Intervalo de confianza; HD-Dex = Dexametasona en dosis alta; IRAC = Comité de Adjudicación de Revisión Independiente; LD-Dex = Dexametasona en dosis baja. a La mediana se basa en la estimación de Kaplan-Meier. b Según el modelo de riesgos proporcionales de Cox que compara las funciones de riesgo asociadas con los grupos de tratamiento, estratificado por edad (≤75 frente a >75), población de enfermedades (refractaria tanto a Lenalidomida como a Bortezomib frente a no refractaria a ambos fármacos) y número previo de terapias anti-mieloma (=2 frente a >2), factores de estratificación para el ensayo. c El valor de p se basa en una prueba de log-rank estratificada con los mismos factores de estratificación que el modelo de Cox anterior. d El 53% de los pacientes del grupo de dosis alta de Dex recibieron posteriormente POMALYST. e Según el modelo de riesgos proporcionales de Cox (no estratificado) que compara las funciones de riesgo asociadas con los grupos de tratamiento. f El valor de p se basa en una prueba de log-rank no estratificada. g Control alfa para SLP y SG. Fecha de corte de datos: 07 de septiembre de 2012 para SLP Fecha de corte de datos: 01 de marzo de 2013 para SG y TRG |
||
|
POMALYST + dosis baja de Dex |
Dosis alta de Dex |
|
|
(N=302) |
(N=153) |
|
|
Tiempo de supervivencia libre de progresión |
||
|
Número (%) de eventos |
164 (54.3) |
103 (67.3) |
|
Medianaa (IC del 95% bilateral) (meses) |
3.6 [3.0, 4.6] |
1.8 [1.6, 2.1] |
|
Hazard Ratio (Pom+LD-Dex:HD-Dex) IC del 95% bilateralb |
0.45 [0.35, 0.59] |
|
|
Prueba de log-rank Valor de p bilateralc |
<0.001 |
|
|
Tiempo de supervivencia globald |
||
|
Número (%) de muertes |
147 (48.7) |
86 (56.2) |
|
Medianaa (IC del 95% bilateral) (meses) |
12.4 [10.4, 15.3] |
8.0 [6.9, 9.0] |
|
Hazard Ratio (Pom+LD-Dex:HD-Dex) IC del 95% bilaterale |
0.70 [0.54, 0.92] |
|
|
Prueba de log-rank Valor de p bilateral f, g |
0.009 |
|
|
Tasa de respuesta global, n (%) |
71 (23.5) |
6 (3.9) |
|
Respuesta completa |
1 (0.3) |
0 |
|
Muy buena respuesta parcial |
8 (2.6) |
1 (0.7) |
|
Respuesta parcial |
62 (20.5) |
5 (3.3) |
Figura 1: Supervivencia Libre de Progresión Con Base en la Revisión de Respuesta de IRAC Según los Criterios de IMWG (Prueba de Rango Logarítmico Stratificado) (Población ITT)
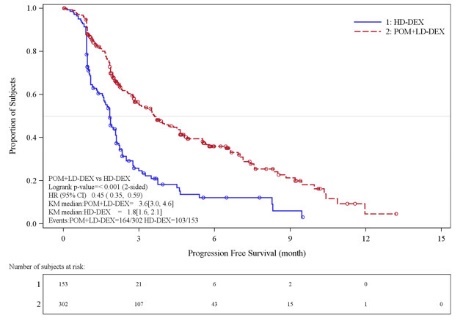
Corte de datos: 07 Sep 2012
Figura 2: Curva de Kaplan-Meier de Supervivencia Global (Población ITT)
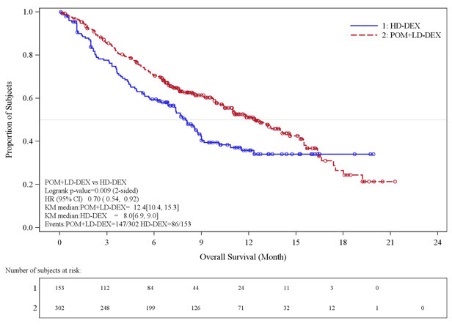
Corte de datos: 01 Mar 2013
14.2 Sarcoma de Kaposi
El ensayo clínico 12-C-0047 (NCT01495598), fue un estudio clínico abierto, de un solo centro y brazo único que evaluó la seguridad y la eficacia de POMALYST en pacientes con sarcoma de Kaposi (KS). Un total de 28 pacientes (18 VIH positivo, 10 VIH negativo) recibieron POMALYST 5 mg por vía oral una vez al día durante los días 1 al 21 de cada ciclo de 28 días hasta la progresión de la enfermedad o toxicidad inaceptable. Todos los pacientes VIH positivo continuaron la terapia antirretroviral altamente activa (HAART). El ensayo excluyó a los pacientes con KS pulmonar o visceral sintomático, historia de tromboembolia venosa o arterial o trastornos procoagulantes. Los pacientes recibieron profilaxis trombótica con aspirina 81 mg una vez al día durante todo el tratamientoo.
La edad mediana era de 52,5 años, todos eran hombres, el 75% eran blancos y el 14% negros o afroamericanos. El 75% de los pacientes tenían enfermedad avanzada (T1) al momento de la inscripción, el 11% tenían ≥ 50 lesiones y el 75% habían recibido quimioterapia previa.
La principal medida de resultados de eficacia era la tasa de respuesta global (ORR), que incluía la respuesta completa (CR), respuesta clínica completa (cCR) y respuesta parcial (PR). La respuesta fue evaluada por el investigador de acuerdo con los criterios de respuesta del Comité Oncológico del Grupo de Ensayos Clínicos del SIDA (ACTG) para KS. El tiempo medio a la primera respuesta fue de 1,8 meses (0,9 a 7,6). Los resultados de eficacia se presentan en la Tabla 11.
| CI: intervalo de confianza, ORR: tasa de respuesta global, CR: respuesta completa, PR: respuesta parcial 1 CR incluye a un paciente VIH negativo que alcanzó una cCR. 2 Calculado como la fecha de la primera respuesta documentada a la fecha de la primera progresión documentada de la enfermedad, la recepción de un nuevo tratamientoo o un segundo curso de tratamientoo, o la muerte por cualquier causa, la que ocurra primero. La estimación mediana es de un análisis de Kaplan-Meier. 3 Del análisis de Kaplan-Meier. |
|||
|
Todos los pacientes |
VIH positivo |
VIH negativo |
|
|
ORR 1, n (%) |
20 (71) |
12 (67) |
8 (80) |
|
[95% CI] |
[51, 87] |
(41, 87) |
(44, 98) |
|
CR 1, n (%) |
4 (14) |
3 (17) |
1 (10) |
|
PR, n (%) |
16 (57) |
9 (50) |
7 (70) |
|
Duración de la Respuesta, KS 2, |
12,1 |
12,5 |
10,5 |
|
Mediana en meses [95% CI]3 |
[7,6, 16,8] |
[6,5, 24,9] |
[3,9, 24,2] |
|
Duración de la Respuesta, KS (%) |
|||
|
Porcentaje mayor de 12 meses |
50 |
58 |
38 |
|
Porcentaje mayor de 24 meses |
20 |
17 |
25 |
15 REFERENCIAS
- 1.
- OSHA Hazardous Drugs. OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Cápsula de tapa opaca azul oscuro y cuerpo opaco amarillo, con la inscripción “POML” en la tapa en tinta blanca y “1 mg” en el cuerpo en tinta negra
|
Frascos de 1 mg de 21 |
(NDC 59572-501-21) |
|
|
Frascos de 1 mg de 100 |
(NDC 59572-501-00) |
Cápsula de tapa opaca azul oscuro y cuerpo opaco naranja, con la inscripción “POML” en la tapa y “2 mg” en el cuerpo en tinta blanca
|
Frascos de 2 mg de 21 |
(NDC 59572-502-21) |
|
|
Frascos de 2 mg de 100 |
(NDC 59572-502-00) |
Cápsula de tapa opaca azul oscuro y cuerpo opaco verde, con la inscripción “POML” en la tapa y “3 mg” en el cuerpo en tinta blanca
|
Frascos de 3 mg de 21 |
(NDC 59572-503-21) |
|
|
Frascos de 3 mg de 100 |
(NDC 59572-503-00) |
Cápsula de tapa opaca azul oscuro y cuerpo opaco azul, con la inscripción “POML” en la tapa y “4 mg” en el cuerpo en tinta blanca
|
Frascos de 4 mg de 21 |
(NDC 59572-504-21) |
|
|
Frascos de 4 mg de 100 |
(NDC 59572-504-00) |
Almacenar a 20°C-25°C (68°F-77°F); se permiten excursiones de 15°C-30°C (59°F-86°F) [ver Temperatura Controlada USP].
Se debe tener cuidado al manipular POMALYST. No abrir ni triturar las cápsulas de POMALYST. Si el polvo de POMALYST entra en contacto con la piel, lavar inmediata y completamente con agua y jabón. Si POMALYST entra en contacto con las membranas mucosas, enjuagar completamente con agua.
Seguir los procedimientos para la manipulación y eliminación adecuada de medicamentos peligrosos. 1
17 INFORMACIÓN PARA EL PACIENTE
Aconseje al paciente que lea la información para pacientes aprobada por la FDA (Guía del Medicamento).
Toxicidad embriofetal
Informe a las pacientes que POMALYST está contraindicado durante el embarazo [consulte Contraindicaciones (4)]. POMALYST es un análogo de la talidomida y puede causar defectos de nacimiento graves o la muerte de un bebé en desarrollo [consulte Advertencias y precauciones (5.1) y Uso en poblaciones específicas (8.1)].
- •
- Aconseje a las mujeres con potencial reproductivo que deben evitar el embarazo mientras toman POMALYST y durante al menos 4 semanas después de completar el tratamiento.
- •
- Inicie el tratamiento con POMALYST en mujeres con potencial reproductivo solo después de una prueba de embarazo negativa.
- •
- Aconseje a las mujeres con potencial reproductivo sobre la importancia de las pruebas de embarazo mensuales y la necesidad de utilizar 2 formas diferentes de anticoncepción, incluida al menos 1 forma altamente eficaz, simultáneamente durante el tratamiento con POMALYST, durante las interrupciones de la dosis y durante 4 semanas después de que haya terminado de tomar POMALYST por completo. Las formas altamente eficaces de anticoncepción, además de la ligadura de trompas, incluyen el DIU y los métodos hormonales (píldoras anticonceptivas, inyecciones, parches o implantes) y la vasectomía de la pareja. Los métodos anticonceptivos eficaces adicionales incluyen el condón de látex o sintético, el diafragma y el capuchón cervical.
- •
- Indique a la paciente que deje de tomar POMALYST inmediatamente y que se ponga en contacto con su proveedor de atención médica si queda embarazada mientras toma este medicamento, si se retrasa su período menstrual o experimenta un sangrado menstrual inusual, si deja de tomar anticonceptivos o si cree POR CUALQUIER MOTIVO que puede estar embarazada.
- •
- Aconseje a la paciente que si su proveedor de atención médica no está disponible, debe llamar al Centro de llamadas REMS al 1-888-423-5436 [consulte Advertencias y precauciones (5.1) y Uso en poblaciones específicas (8.3)].
- •
- Aconseje a los hombres que siempre usen un condón de látex o sintético durante cualquier contacto sexual con mujeres con potencial reproductivo mientras toman POMALYST y hasta por 4 semanas después de suspender POMALYST, incluso si se han sometido a una vasectomía exitosa.
- •
- Aconseje a los pacientes varones que toman POMALYST que no deben donar esperma [consulte Advertencias y precauciones (5.1) y Uso en poblaciones específicas (8.3)].
- •
- Se debe indicar a todos los pacientes que no donen sangre mientras toman POMALYST y durante 4 semanas después de la suspensión de POMALYST [consulte Advertencias y precauciones (5.1)].
Programa REMS de POMALYST
Debido al riesgo de toxicidad embriofetal, POMALYST solo está disponible a través de un programa restringido llamado POMALYST REMS [consulte Advertencias y precauciones (5.2)].
- •
- Los pacientes deben firmar un formulario de acuerdo entre el paciente y el médico y cumplir con los requisitos para recibir POMALYST. En particular, las mujeres con potencial reproductivo deben cumplir con los requisitos de las pruebas de embarazo y la anticoncepción, y participar en encuestas telefónicas mensuales. Los hombres deben cumplir con los requisitos de anticoncepción [consulte Uso en poblaciones específicas (8.3)].
- •
- POMALYST solo está disponible en farmacias certificadas en el programa POMALYST REMS. Proporcione a los pacientes el número de teléfono y el sitio web para obtener información sobre cómo obtener el producto.
Registro de exposición durante el embarazo
Informe a las mujeres que existe un Registro de exposición durante el embarazo que monitorea los resultados del embarazo en mujeres expuestas a POMALYST durante el embarazo y que pueden comunicarse con el Registro de exposición durante el embarazo llamando al 1-888-423-5436 [consulte Uso en poblaciones específicas (8.1)].
Tromboembolia venosa y arterial
Informe a los pacientes sobre el riesgo de desarrollar TVP, EP, IM y accidente cerebrovascular, y que informen inmediatamente cualquier signo y síntoma que sugiera estos eventos para su evaluación [consulte Advertencias y precauciones (5.3)].
Toxicidades hematológicas
Informe a los pacientes sobre los riesgos de desarrollar neutropenia, trombocitopenia y anemia, y la necesidad de informar los signos y síntomas asociados con estos eventos a su proveedor de atención médica para una evaluación adicional [consulte Advertencias y precauciones (5.5)].
Hepatotoxicidad
Informe a los pacientes sobre los riesgos de desarrollar hepatotoxicidad, incluida la insuficiencia hepática y la muerte, y que informen los signos y síntomas asociados con estos eventos a su proveedor de atención médica para su evaluación [consulte Advertencias y precauciones (5.6)].
Reacciones cutáneas graves
Informe a los pacientes sobre el riesgo potencial de reacciones cutáneas graves, como SSJ, NET y DRESS, y que informen cualquier signo y síntoma asociado con estas reacciones a su proveedor de atención médica para su evaluación [consulte Advertencias y precauciones (5.7)].
Mareos y estado confusional
Informe a los pacientes sobre el riesgo potencial de mareos y estado confusional con el medicamento, que eviten situaciones en las que los mareos o el estado confusional puedan ser un problema, y que no tomen otros medicamentos que puedan causar mareos o estado confusional sin el consejo médico adecuado [consulte Advertencias y precauciones (5.8)].
Neuropatía
Informe a los pacientes sobre el riesgo de neuropatía y pídales que informen a su proveedor de atención médica sobre los signos y síntomas asociados con estos eventos para una evaluación adicional [consulte Advertencias y precauciones (5.9)].
Segundas neoplasias malignas primarias
Informe al paciente que se desconoce el riesgo potencial de desarrollar leucemia mieloide aguda durante el tratamiento con POMALYST [consulte Advertencias y precauciones (5.10)].
Síndrome de lisis tumoral
Informe a los pacientes sobre el riesgo potencial de síndrome de lisis tumoral y pídales que informen cualquier signo y síntoma asociado con este evento a su proveedor de atención médica para su evaluación [consulte Advertencias y precauciones (5.11)].
Hipersensibilidad
Informe a los pacientes sobre la posibilidad de reacciones de hipersensibilidad graves, como angioedema y anafilaxia a POMALYST. Indique a los pacientes que se comuniquen con su proveedor de atención médica de inmediato si presentan cualquier signo y síntoma de estas reacciones. Aconseje a los pacientes que busquen atención médica de emergencia si presentan signos o síntomas de reacciones de hipersensibilidad graves [consulte Advertencias y precauciones (5.12)].
Consumo de tabaco
Aconseje a los pacientes que fumar tabaco puede reducir la eficacia de POMALYST [consulte Uso en poblaciones específicas (8.8) y Farmacología clínica (12.3)].
Instrucciones de dosificación
Informe a los pacientes sobre cómo tomar POMALYST [consulte Dosificación y administración (2.2, 2.3, 2.9)]
- •
- POMALYST debe tomarse una vez al día, aproximadamente a la misma hora todos los días.
- •
- Los pacientes en hemodiálisis deben tomar POMALYST después de la hemodiálisis, los días de hemodiálisis.
- •
- POMALYST puede tomarse con o sin alimentos.
- •
- Las cápsulas no deben abrirse, romperse ni masticarse. POMALYST debe tragarse entero con agua.
- •
- Indique a los pacientes que si olvidan una dosis de POMALYST, aún pueden tomarla hasta 12 horas después de la hora en que normalmente la tomarían. Si han transcurrido más de 12 horas, se les debe indicar que omitan la dosis de ese día. Al día siguiente, deben tomar POMALYST a la hora habitual. Advierta a los pacientes que no tomen 2 dosis para compensar la que olvidaron.
SECCIÓN NO CLASIFICADA SPL
Comercializado por: Bristol-Myers Squibb Company
Princeton, NJ 08543 USA
POMALYST® y POMALYST REMS® son marcas comerciales de Celgene Corporation, una compañía de Bristol-Myers Squibb.
POMPI.016/MG.014
Guía de medicación
|
Esta Guía del Medicamento ha sido aprobada por la Administración de Alimentos y Medicamentos de los Estados Unidos. |
Revisado: Marzo 2023 |
|||
|
GUÍA DEL MEDICAMENTO |
||||
|
¿Cuál es la información más importante que debo saber sobre POMALYST? |
||||
|
Antes de comenzar a tomar POMALYST, debe leer y aceptar todas las instrucciones del programa POMALYST REMS®. Para obtener más información, llame al 1-888-423-5436 o visite www.pomalystrems.com. Antes de recetarle POMALYST, su proveedor de atención médica le explicará el programa POMALYST REMS y le hará firmar el Formulario de Acuerdo Paciente-Médico. |
||||
|
POMALYST puede causar efectos secundarios graves, que incluyen: |
||||
|
||||
|
¿Qué es POMALYST? |
||||
|
POMALYST es un medicamento recetado que se usa para tratar a adultos con: |
||||
|
||||
|
No se sabe si POMALYST es seguro y eficaz en niños. |
||||
|
¿Quién no debe tomar POMALYST? |
||||
|
No tome POMALYST si usted: |
||||
|
||||
|
¿Qué debo decirle a mi proveedor de atención médica antes de tomar POMALYST? |
||||
|
Antes de tomar POMALYST, informe a su proveedor de atención médica si usted: |
||||
|
||||
|
Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos herbales. POMALYST y otros medicamentos pueden afectar entre sí, causando efectos secundarios graves. Hable con su proveedor de atención médica antes de tomar cualquier medicamento nuevo. |
||||
|
Conozca los medicamentos que toma. Mantenga una lista de ellos para mostrársela a su proveedor de atención médica y farmacéutico. |
||||
|
¿Cómo debo tomar POMALYST?
|
||||
|
¿Qué debo evitar mientras tomo POMALYST? |
||||
|
||||
|
¿Cuáles son los posibles efectos secundarios de POMALYST? |
||||
|
POMALYST puede causar efectos secundarios graves, que incluyen: |
||||
|
||||
|
|
|||
|
||||
|
|
|
||
|
Busque atención médica de emergencia de inmediato si desarrolla alguno de los siguientes signos o síntomas durante el tratamiento con POMALYST: |
||||
|
|
|||
|
||||
|
Su proveedor de atención médica puede decirle que deje de tomar POMALYST si desarrolla ciertos efectos secundarios graves durante el tratamiento. |
||||
|
Los efectos secundarios más comunes de POMALYST en personas con mieloma múltiple incluyen: |
||||
|
|
|
||
|
Los efectos secundarios más comunes de POMALYST en personas con KS incluyen: |
||||
|
|
|||
|
Estos no son todos los posibles efectos secundarios de POMALYST. |
||||
|
Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088. |
||||
|
¿Cómo debo almacenar POMALYST? |
||||
|
||||
|
Mantenga POMALYST y todos los medicamentos fuera del alcance de los niños. |
||||
|
Información general sobre el uso seguro y eficaz de POMALYST. |
||||
|
Los medicamentos a veces se recetan para fines distintos de los que se enumeran en una Guía de medicamentos. No tome POMALYST para afecciones para las que no se lo recetaron. No le dé POMALYST a otras personas, incluso si tienen los mismos síntomas que usted. Puede hacerles daño y puede causar defectos de nacimiento. |
||||
|
Si desea obtener más información, hable con su proveedor de atención médica. Puede pedirle a su proveedor de atención médica o farmacéutico información sobre POMALYST que esté escrita para profesionales de la salud. |
||||
|
Para obtener más información, llame al 1-888-423-5436 o visite www.pomalystrems.com. |
||||
|
¿Cuáles son los ingredientes de POMALYST? |
||||
|
Ingrediente activo: pomalidomida |
||||
|
Ingredientes inactivos: manitol, almidón pregelatinizado y estearato de sodio. |
||||
|
La cápsula de 1 mg contiene gelatina, dióxido de titanio, FD&C azul 2, óxido de hierro amarillo, tinta blanca y tinta negra. |
||||
|
Comercializado por: Bristol-Myers Squibb Company, Princeton, NJ 08543 EE. UU. |
||||
Envase representativo de POMALYST 1 mg
NDC 59572-501-21
Pomalyst®
(pomalidomida) cápsulas
1 mg
ADVERTENCIA: POTENCIAL PARA DEFECTOS CONGÉNITOS HUMANOS.
Sólo con receta
21 Cápsulas
Bristol Myers Squibb

Envase representativo de POMALYST 2 mg
NDC 59572-502-21
Pomalyst®
(pomalidomida) cápsulas
2 mg
ADVERTENCIA: POTENCIAL PARA DEFECTOS CONGÉNITOS HUMANOS.
Receta médica solamente
21 Cápsulas
Bristol Myers Squibb

Envase representativo de POMALYST 3 mg
NDC 59572-503-21
Pomalyst®
(pomalidomida) cápsulas
3 mg
ADVERTENCIA: POTENCIAL PARA DEFECTOS CONGÉNITOS HUMANOS.
Únicamente con receta
21 Cápsulas
Bristol Myers Squibb

Envase representativo de POMALYST 4 mg
NDC 59572-504-21
Pomalyst®
(pomalidomida) cápsulas
4 mg
ADVERTENCIA: POTENCIAL DE DEFECTOS CONGÉNITOS HUMANOS.
Únicamente con receta
21 Cápsulas
Bristol Myers Squibb
