ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
Estos aspectos destacados no incluyen toda la información necesaria para usar REVLIMID® de forma segura y eficaz. Consulte la información de prescripción completa para REVLIMID.
REVLIMID (lenalidomida) cápsulas, para administración oral Aprobación inicial en EE. UU.: 2005
ADVERTENCIA: TOXICIDAD EMBRIOFETAL, TOXICIDAD HEMATOLÓGICA Y TROMBOEMBOLISMO VENOSO Y ARTERIAL
Consulte la información de prescripción completa para obtener la advertencia completa en recuadro.
TOXICIDAD EMBRIOFETAL
•
La lenalidomida, un análogo de la talidomida, causó anomalías en las extremidades en un estudio de desarrollo en monos similar a los defectos de nacimiento causados por la talidomida en humanos. Si se usa lenalidomida durante el embarazo, puede causar defectos de nacimiento o muerte embrionaria o fetal.
•
Se debe excluir el embarazo antes de comenzar el tratamiento. Evite el embarazo durante el tratamiento mediante el uso de dos métodos anticonceptivos confiables (5.1).
REVLIMID solo está disponible a través de un programa de distribución restringido, llamado programa Lenalidomide REMS (5.2, 17).
TOXICIDAD HEMATOLÓGICA.REVLIMID puede causar neutropenia y trombocitopenia significativas (5.3).
TROMBOEMBOLISMO VENOSO Y ARTERIAL
Riesgo significativamente mayor de trombosis venosa profunda (TVP) y embolia pulmonar (EP), así como riesgo de infarto de miocardio y accidente cerebrovascular en pacientes con mieloma múltiple que reciben REVLIMID con dexametasona. Se recomienda la profilaxis antitrombótica (5.4).
REVLIMID es un análogo de la talidomida indicado para el tratamiento de pacientes adultos con:
•
Mieloma múltiple (MM), en combinación con dexametasona (1.1).
•
MM, como mantenimiento después del trasplante autólogo de células madre hematopoyéticas (auto-HSCT) (1.1).
•
Anemia dependiente de transfusiones debida a síndromes mielodisplásicos de riesgo bajo o intermedio-1 asociados a una anomalía de deleción 5q, con o sin anomalías citogenéticas adicionales (1.2).
•
Linfoma de células de la capa manto (MCL) cuya enfermedad ha reaparecido o progresado después de dos terapéuticas previas, una de las cuales incluyó bortezomib (1.3).
•
Linfoma folicular tratado previamente (FL), en combinación con un producto de rituximab (1.4).
•
Linfoma de zona marginal tratado previamente (MZL), en combinación con un producto de rituximab (1.5).
Limitaciones de uso:
•
REVLIMID no está indicado y no se recomienda para el tratamiento de pacientes con leucemia linfocítica crónica (CLL) fuera de ensayos clínicos controlados (1.4).
DOSIFICACIÓN Y ADMINISTRACIÓN
•
Terapia de combinación para MM: 25 mg una vez al día por vía oral en los días 1-21 de ciclos repetidos de 28 días. (2.1).
•
Terapia de mantenimiento de MM después de auto-HSCT: 10 mg una vez al día continuamente en los días 1-28 de ciclos repetidos de 28 días (2.1).
Hiperalergia severa demostrada a la lenalidomida (4.2, 5.9, 5.15).
ADVERTENCIAS Y PRECAUCIONES
•
Mortalidad aumentada: Reacciones adversas cardiacas graves y fatales ocurrieron en pacientes con CLL tratados con REVLIMID (5.5).
•
Segundos cánceres primarios (SPM): Incidencias más altas de SPM se observaron en ensayos controlados de pacientes con MM que recibieron REVLIMID (5.6).
•
Mortalidad aumentada: Observada en pacientes con MM cuando pembrolizumab se agregó a dexametasona y un análogo de talidomida (5.7).
•
Hepatotoxicidad: Insuficiencia hepática incluyendo fatalidades; monitorear la función hepática. Detener REVLIMID y evaluar si se sospecha hepatotoxicidad (5.8).
•
Reacciones cutáneas graves: Suspender REVLIMID para reacciones graves (5.9).
•
Síndrome de lisis tumoral (TLS) incluyendo fatalidades: Monitorear pacientes en riesgo de TLS (es decir, aquellos con alta carga tumoral) y tomar las precauciones adecuadas (5.10).
•
Reacción de llamarada tumoral: Reacciones de llamarada tumoral graves, incluyendo reacciones fatales, han ocurrido durante el uso investigativo de REVLIMID para leucemia linfocítica crónica y linfoma (5.11).
•
Impedimento en la movilización de células madre: Se ha reportado una disminución en el número de células CD34+ recogidas después del tratamiento ( > 4 ciclos) con REVLIMID. Considerar la remisión temprana al centro de trasplante (5.12).
•
Mortalidad temprana en MCL: Una mayor tasa de muertes tempranas ha ocurrido en pacientes con MCL (5.14).
•
Hiperalergia: Monitorear pacientes por potencial hiperalergia. Suspender REVLIMID para angioedema y anafilaxia (5.15).
REACCIONES ADVERSAS
•
MM: Las reacciones adversas más comunes (≥20%) incluyen diarrea, fatiga, anemia, estreñimiento, neutropenia, leucopenia, edema periférico, insomnio, calambres/espasmos musculares, dolor abdominal, dolor de espalda, náuseas, astenia, fiebre, infección de las vías respiratorias superiores, bronquitis, nasofaringitis, gastroenteritis, tos, erupción cutánea, disnea, mareo, pérdida de apetito, trombocitopenia y temblor (6.1).
•
MDS: Las reacciones adversas más comunes ( > 15%) incluyen trombocitopenia, neutropenia, diarrea, prurito, erupción cutánea, fatiga, estreñimiento, náuseas, nasofaringitis, artralgia, fiebre, dolor de espalda, edema periférico, tos, mareo, dolor de cabeza, calambres musculares, disnea, faringitis y epistaxis (6.1).
•
Linfoma no Hodgkin (NHL: MCL, FL o MZL): Las reacciones adversas más comunes (≥15%) incluyen neutropenia, trombocitopenia, anemia, leucopenia, diarrea, estreñimiento, náuseas, fatiga, fiebre, tos, infección de las vías respiratorias superiores y erupción cutánea (6.1).
Para reportar REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Bristol-Myers Squibb al 1-800-721-5072 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
INTERACCIONES MEDICAMENTOSAS
•
Digoxina: Monitorear los niveles plasmáticos de digoxina periódicamente debido al aumento de Cmax y AUC con la terapia concomitante con REVLIMID (7.1).
•
El uso concomitante de agentes estimulantes de la eritropoyetina o terapias que contienen estrógenos con REVLIMID puede aumentar el riesgo de trombosis (7.2).
Las secciones o subsecciones omitidas de la información completa de prescripción no están enumeradas.
ADVERTENCIA EN LA CAJA
ADVERTENCIA: TOXICIDAD EMBRIOFETAL, TOXICIDAD HEMATOLÓGICA Y TROMBOEMBOLISMO VENOSO Y ARTERIAL
Toxicidad embrio-fetal
No use REVLIMID durante el embarazo. La lenalidomida, un análogo de la talidomida, causó anomalías en las extremidades en un estudio de desarrollo en monos. La talidomida es un teratógeno humano conocido que causa defectos de nacimiento humanos graves que ponen en peligro la vida. Si se usa lenalidomida durante el embarazo, puede causar defectos de nacimiento o muerte embrio-fetal. En mujeres en edad fértil, obtenga 2 pruebas de embarazo negativas antes de comenzar el tratamiento con REVLIMID®. Las mujeres en edad fértil deben usar 2 métodos anticonceptivos o abstenerse continuamente de las relaciones sexuales heterosexuales durante y durante las 4 semanas posteriores al tratamiento con REVLIMID [ver Advertencias y precauciones (5.1) y Guía de medicamentos (17)]. Para evitar la exposición embrio-fetal a la lenalidomida, REVLIMID solo está disponible a través de un programa de distribución restringido, el programa Lenalidomide REMS (5.2).
Información sobre el programa Lenalidomide REMS está disponible en www.lenalidomiderems.com o llamando al Centro de llamadas de REMS al 1-888-423-5436.
Toxicidad hematológica (neutropenia y trombocitopenia)
REVLIMID puede causar neutropenia y trombocitopenia significativas. El ochenta por ciento de los pacientes con síndromes mielodisplásicos del 5q tuvieron que retrasar/reducir la dosis durante el estudio principal. El treinta y cuatro por ciento de los pacientes tuvieron que retrasar/reducir la dosis por segunda vez. La toxicidad hematológica de grado 3 o 4 se observó en el 80% de los pacientes inscritos en el estudio. Los pacientes en terapia para síndromes mielodisplásicos del 5q deben controlar su hemograma completo semanalmente durante las primeras 8 semanas de terapia y al menos mensualmente a partir de entonces. Los pacientes pueden requerir interrupción y/o reducción de la dosis. Los pacientes pueden requerir el uso de apoyo de productos sanguíneos y/o factores de crecimiento [ver Dosificación y administración (2.2)].
Tromboembolismo venoso y arterial
REVLIMID ha demostrado un riesgo significativamente mayor de trombosis venosa profunda (TVP) y embolia pulmonar (EP), así como riesgo de infarto de miocardio y accidente cerebrovascular en pacientes con mieloma múltiple que fueron tratados con REVLIMID y terapia con dexametasona. Monitoree y asesore a los pacientes sobre los signos y síntomas de tromboembolismo. Avise a los pacientes que busquen atención médica inmediata si desarrollan síntomas como dificultad para respirar, dolor en el pecho o hinchazón en los brazos o las piernas. Se recomienda la tromboprofilaxis y la elección del régimen debe basarse en una evaluación de los riesgos subyacentes del paciente [ver Advertencias y precauciones (5.4)].
1 INDICACIONES Y USO
1.1 Mieloma Múltiple
REVLIMID en combinación con dexametasona está indicado para el tratamiento de pacientes adultos con mieloma múltiple (MM).
REVLIMID está indicado como terapia de mantenimiento en pacientes adultos con MM después del trasplante autólogo de células madre hematopoyéticas (auto-HSCT).
1.2 Síndromes Mielodisplásicos
REVLIMID está indicado para el tratamiento de pacientes adultos con anemia dependiente de transfusiones debido a síndromes mielodisplásicos (SMD) de bajo o intermedio-1 riesgo asociados con una anomalía citogenética de deleción 5q con o sin anomalías citogenéticas adicionales.
1.3 Linfoma de Células del Manto
REVLIMID está indicado para el tratamiento de pacientes adultos con linfoma de células del manto (LCM) cuya enfermedad ha recaído o progresado después de dos terapias previas, una de las cuales incluyó bortezomib.
1.4 Linfoma Folicular
REVLIMID en combinación con un producto de rituximab, está indicado para el tratamiento de pacientes adultos con linfoma folicular (LF) previamente tratado.
1.5 Linfoma de Zona Marginal
REVLIMID en combinación con un producto de rituximab, está indicado para el tratamiento de pacientes adultos con linfoma de zona marginal (LZM) previamente tratado.
1.6 Limitaciones de Uso
REVLIMID no está indicado y no se recomienda para el tratamiento de pacientes con CLL fuera de ensayos clínicos controlados [ver Advertencias y Precauciones (5.5)].
2 DOSIS Y ADMINISTRACIÓN
2.1 Dosis recomendada para el mieloma múltiple
Terapia combinada con REVLIMID
La dosis inicial recomendada de REVLIMID es de 25 mg por vía oral una vez al día los días 1 a 21 de ciclos repetidos de 28 días en combinación con dexametasona. Consulte la Sección 14.1 para conocer la dosificación específica de dexametasona. En pacientes mayores de 75 años, la dosis inicial de dexametasona puede reducirse [consulte Estudios clínicos (14.1)]. El tratamiento debe continuarse hasta la progresión de la enfermedad o una toxicidad inaceptable.
En pacientes que no son elegibles para auto-HSCT, el tratamiento debe continuarse hasta la progresión de la enfermedad o una toxicidad inaceptable. Para los pacientes elegibles para auto-HSCT, la movilización de células madre hematopoyéticas debe ocurrir dentro de los 4 ciclos de una terapia que contenga REVLIMID [consulte Advertencias y precauciones (5.12)].
Ajustes de dosis para toxicidades hematológicas durante el tratamiento del MM
Se recomienda seguir las pautas de modificación de la dosis, como se resume en la Tabla 1 a continuación, para controlar la neutropenia o trombocitopenia de grado 3 o 4 u otra toxicidad de grado 3 o 4 que se considere relacionada con REVLIMID.
Tabla 1: Ajustes de dosis para toxicidades hematológicas para MM
Recuentos de plaquetas
Trombocitopenia en MM
Cuando las plaquetas
Curso recomendado
Días 1 a 21 de un ciclo repetido de 28 días
Caen por debajo de 30 000/mcL
Interrumpir el tratamiento con REVLIMID, seguir el CBC semanalmente
Vuelven a al menos 30 000/mcL
Reanudar REVLIMID a la siguiente dosis más baja. No administrar dosis inferiores a 2,5 mg al día
Por cada caída posterior por debajo de 30 000/mcL
Interrumpir el tratamiento con REVLIMID
Vuelven a al menos 30 000/mcL
Reanudar REVLIMID a la siguiente dosis más baja. No administrar dosis inferiores a 2,5 mg al día
Recuentos absolutos de neutrófilos (ANC)
Neutropenia en MM
Cuando los neutrófilos
Curso recomendado
Días 1 a 21 de un ciclo repetido de 28 días
Caen por debajo de 1000/mcL
Interrumpir el tratamiento con REVLIMID, seguir el CBC semanalmente
Vuelven a al menos 1000/mcL y la neutropenia es la única toxicidad
Reanudar REVLIMID a 25 mg al día o la dosis inicial inicial
Vuelven a al menos 1000/mcL y si hay otra toxicidad
Reanudar REVLIMID a la siguiente dosis más baja. No administrar dosis inferiores a 2,5 mg al día
Por cada caída posterior por debajo de 1000/mcL
Interrumpir el tratamiento con REVLIMID
Vuelven a al menos 1000/mcL
Reanudar REVLIMID a la siguiente dosis más baja. No administrar dosis inferiores a 2,5 mg al día
Terapia de mantenimiento con REVLIMID después de un auto-HSCT
Después de un auto-HSCT, inicie la terapia de mantenimiento con REVLIMID después de una recuperación hematológica adecuada (RAN ≥ 1000/mcL y/o recuentos de plaquetas ≥ 75 000/mcL). La dosis inicial recomendada de REVLIMID es de 10 mg una vez al día de forma continua (días 1 a 28 de ciclos repetidos de 28 días) hasta la progresión de la enfermedad o la aparición de una toxicidad inaceptable. Después de 3 ciclos de terapia de mantenimiento, la dosis puede aumentarse a 15 mg una vez al día si se tolera.
Ajustes de dosis para toxicidades hematológicas durante el tratamiento del MM
Se recomiendan las pautas de modificación de la dosis, como se resume en la Tabla 2 a continuación, para controlar la neutropenia o trombocitopenia de Grado 3 o 4 u otra toxicidad de Grado 3 o 4 que se considere relacionada con REVLIMID.
Tabla 2: Ajustes de dosis para toxicidades hematológicas para MM
Recuentos de plaquetas
Trombocitopenia en MM
Cuando las plaquetas
Medidas recomendadas
Caen por debajo de 30 000/mcL
Interrumpir el tratamiento con REVLIMID, realizar un hemograma completo semanalmente
Vuelven a al menos 30 000/mcL
Reanudar REVLIMID a la siguiente dosis más baja, de forma continua durante los días 1 a 28 de un ciclo repetido de 28 días
Si con la dosis diaria de 5 mg, para una caída posterior por debajo de 30 000/mcL
Interrumpir el tratamiento con REVLIMID. No administrar dosis inferiores a 5 mg al día del día 1 al 21 del ciclo de 28 días
Vuelven a al menos 30 000/mcL
Reanudar REVLIMID a 5 mg al día durante los días 1 a 21 del ciclo de 28 días. No administrar dosis inferiores a 5 mg al día del día 1 al 21 del ciclo de 28 días
Recuentos absolutos de neutrófilos (RAN)
Neutropenia en MM
Cuando los neutrófilos
Medidas recomendadas
Caen por debajo de 500/mcL
Interrumpir el tratamiento con REVLIMID, realizar un hemograma completo semanalmente
Vuelven a al menos 500/mcL
Reanudar REVLIMID a la siguiente dosis más baja, de forma continua durante los días 1 a 28 de un ciclo repetido de 28 días
Si con la dosis diaria de 5 mg, para una caída posterior por debajo de 500/mcL
Interrumpir el tratamiento con REVLIMID. No administrar dosis inferiores a 5 mg al día del día 1 al 21 del ciclo de 28 días
Vuelven a al menos 500/mcL
Reanudar REVLIMID a 5 mg al día durante los días 1 a 21 del ciclo de 28 días. No administrar dosis inferiores a 5 mg al día del día 1 al 21 del ciclo de 28 días
2.2 Dosis recomendada para los síndromes mielodisplásicos
La dosis inicial recomendada de REVLIMID es de 10 mg al día. El tratamiento se continúa o modifica en función de los hallazgos clínicos y de laboratorio. Continúe el tratamiento hasta la progresión de la enfermedad o la aparición de una toxicidad inaceptable.
Ajustes de dosis para las toxicidades hematológicas durante el tratamiento de los síndromes mielodisplásicos
En los pacientes que reciben una dosis inicial de 10 mg y que experimentan trombocitopenia, la dosis debe ajustarse de la siguiente manera:
Recuentos de plaquetas
Si la trombocitopenia se desarrolla DENTRO de las 4 semanas de iniciar el tratamiento con 10 mg diarios para los síndromes mielodisplásicos
Si el valor inicial es de al menos 100 000/mcL
Cuando las plaquetas
Medidas recomendadas
Desciendan por debajo de 50 000/mcL
Interrumpir el tratamiento con REVLIMID
Vuelvan a ser de al menos 50 000/mcL
Reanudar REVLIMID a 5 mg al día
Si el valor inicial es inferior a 100 000/mcL
Cuando las plaquetas
Medidas recomendadas
Desciendan al 50 % del valor inicial
Interrumpir el tratamiento con REVLIMID
Si el valor inicial es de al menos 60 000/mcL y vuelven a ser de al menos 50 000/mcL
Reanudar REVLIMID a 5 mg al día
Si el valor inicial es inferior a 60 000/mcL y vuelven a ser de al menos 30 000/mcL
Reanudar REVLIMID a 5 mg al día
Si la trombocitopenia se desarrolla DESPUÉS de 4 semanas de iniciar el tratamiento con 10 mg diarios para los síndromes mielodisplásicos
Cuando las plaquetas
Medidas recomendadas
Desciendan por debajo de 30 000/mcL o por debajo de 50 000/mcL con transfusiones de plaquetas
Interrumpir el tratamiento con REVLIMID
Vuelvan a ser de al menos 30 000/mcL (sin insuficiencia hemostática)
Reanudar REVLIMID a 5 mg al día
En los pacientes que experimentan trombocitopenia con 5 mg diarios, la dosis debe ajustarse de la siguiente manera:
Si la trombocitopenia se desarrolla durante el tratamiento con 5 mg diarios para los síndromes mielodisplásicos
Cuando las plaquetas
Medidas recomendadas
Desciendan por debajo de 30 000/mcL o por debajo de 50 000/mcL con transfusiones de plaquetas
Interrumpir el tratamiento con REVLIMID
Vuelvan a ser de al menos 30 000/mcL (sin insuficiencia hemostática)
Reanudar REVLIMID a 2.5 mg al día
En los pacientes que reciben una dosis inicial de 10 mg y que experimentan neutropenia, la dosis debe ajustarse de la siguiente manera:
Recuentos absolutos de neutrófilos (RAN)
Si la neutropenia se desarrolla DENTRO de las 4 semanas de iniciar el tratamiento con 10 mg diarios para los síndromes mielodisplásicos
Si el RAN inicial es de al menos 1000/mcL
Cuando los neutrófilos
Medidas recomendadas
Desciendan por debajo de 750/mcL
Interrumpir el tratamiento con REVLIMID
Vuelvan a ser de al menos 1000/mcL
Reanudar REVLIMID a 5 mg al día
Si el RAN inicial es inferior a 1000/mcL
Cuando los neutrófilos
Medidas recomendadas
Desciendan por debajo de 500/mcL
Interrumpir el tratamiento con REVLIMID
Vuelvan a ser de al menos 500/mcL
Reanudar REVLIMID a 5 mg al día
Si se desarrolla neutropenia DESPUÉS de 4 semanas de comenzar el tratamiento con 10 mg diarios en MDS
Cuando los neutrófilos
Curso recomendado
Caen por debajo de 500/mcL durante al menos 7 días o por debajo de 500/mcL asociado con fiebre (al menos 38.5°C)
Interrumpir el tratamiento con REVLIMID
Vuelven a al menos 500/mcL
Reanudar REVLIMID a 5 mg diarios
Los pacientes que experimentan neutropenia con 5 mg diarios deben tener su dosis ajustada de la siguiente manera:
Si se desarrolla neutropenia durante el tratamiento con 5 mg diarios en MDS
Cuando los neutrófilos
Curso recomendado
Caen por debajo de 500/mcL durante al menos 7 días o por debajo de 500/mcL asociado con fiebre (al menos 38.5°C)
Interrumpir el tratamiento con REVLIMID
Vuelven a al menos 500/mcL
Reanudar REVLIMID a 2.5 mg diarios
2.3 Dosis recomendada para el linfoma de células del manto
La dosis inicial recomendada de REVLIMID es de 25 mg/día por vía oral en los días 1 a 21 de ciclos repetidos de 28 días para el linfoma de células del manto en recaída o refractario. El tratamiento debe continuarse hasta la progresión de la enfermedad o la toxicidad inaceptable.
El tratamiento se continúa, modifica o suspende en función de los hallazgos clínicos y de laboratorio.
Ajustes de dosis para toxicidades hematológicas durante el tratamiento de MCL
Se recomiendan las pautas de modificación de la dosis que se resumen a continuación para controlar la neutropenia o trombocitopenia de grado 3 o 4 u otras toxicidades de grado 3 o 4 que se consideran relacionadas con REVLIMID.
Recuentos de plaquetas
Trombocitopenia durante el tratamiento de MCL
Cuando las plaquetas
Curso recomendado
Caen por debajo de 50,000/mcL
Interrumpir el tratamiento con REVLIMID y realizar un seguimiento del CBC semanalmente
Vuelven a al menos 50,000/mcL
Reanudar REVLIMID a 5 mg menos que la dosis anterior. No administrar dosis inferiores a 5 mg diarios
Recuentos absolutos de neutrófilos (ANC)
Neutropenia durante el tratamiento de MCL
Cuando los neutrófilos
Curso recomendado
Caen por debajo de 1,000/mcL durante al menos 7 días O Caen por debajo de 1,000/mcL con una temperatura asociada de al menos 38.5°C O Caen por debajo de 500/mcL
Interrumpir el tratamiento con REVLIMID y realizar un seguimiento del CBC semanalmente
Vuelven a al menos 1,000/mcL
Reanudar REVLIMID a 5 mg menos que la dosis anterior. No administrar dosis inferiores a 5 mg diarios
2.4 Dosis recomendada para linfoma folicular o linfoma de zona marginal
La dosis inicial recomendada de REVLIMID es de 20 mg por vía oral una vez al día en los días 1 a 21 de ciclos repetidos de 28 días durante un máximo de 12 ciclos de tratamiento en combinación con un producto de rituximab. Consulte la Sección 14.4 para conocer la dosificación específica de rituximab del ensayo AUGMENT. Para conocer los ajustes de dosis debidos a la toxicidad con rituximab, consulte la información de prescripción del producto.
Ajustes de dosis para toxicidades hematológicas durante el tratamiento de FL o LZM
Se recomiendan las pautas de modificación de la dosis, como se resume a continuación, para controlar la neutropenia o trombocitopenia de grado 3 o 4 u otra toxicidad de grado 3 o 4 que se considere relacionada con REVLIMID.
Recuentos de plaquetas
Trombocitopenia durante el tratamiento de FL o LZM
Cuando las plaquetas
Curso recomendado
Caen por debajo de 50 000/mcL
Interrumpa el tratamiento con REVLIMID y realice un seguimiento del CBC semanalmente.
Vuelven a al menos 50 000/mcL
Si la dosis inicial del paciente era de 20 mg al día, reanude REVLIMID a 5 mg menos que la dosis anterior. No administre dosis inferiores a 5 mg al día. Si la dosis inicial del paciente era de 10 mg al día, reanude a 5 mg menos que la dosis anterior. No administre dosis inferiores a 2,5 mg al día.
Recuentos absolutos de neutrófilos (RAN)
Neutropenia durante el tratamiento de FL o LZM
Cuando los neutrófilos
Curso recomendado
Caen por debajo de 1000/mcL durante al menos 7 días O Caen por debajo de 1000/mcL con una temperatura asociada de al menos 38,5 °C O Caen por debajo de 500/mcL
Interrumpa el tratamiento con REVLIMID y realice un seguimiento del CBC semanalmente.
Vuelven a al menos 1000/mcL
Si la dosis inicial del paciente era de 20 mg al día, reanude REVLIMID a 5 mg menos que la dosis anterior. No administre dosis inferiores a 5 mg al día. Si la dosis inicial del paciente era de 10 mg al día, reanude a 5 mg menos que la dosis anterior. No administre dosis inferiores a 2,5 mg al día.
2.5 Modificaciones de la dosis para reacciones adversas no hematológicas
Para las toxicidades no hematológicas de grado 3/4 que se considere que están relacionadas con REVLIMID, suspenda el tratamiento y reinícielo a discreción del médico en el siguiente nivel de dosis más bajo cuando la toxicidad se haya resuelto a grado 2 o inferior.
Interrumpa permanentemente REVLIMID en caso de angioedema, anafilaxia, erupción cutánea de grado 4, exfoliación cutánea, ampollas o cualquier otra reacción dermatológica grave [consulte Advertencias y precauciones (5.9, 5.15)].
2.6 Dosis recomendada para pacientes con insuficiencia renal
Las recomendaciones para la dosificación de pacientes con insuficiencia renal se muestran en la siguiente tabla [consulte Farmacología clínica (12.3)].
Tabla 3: Ajustes de dosis para pacientes con insuficiencia renal
Función renal
(Cockcroft-Gault)
Dosis en la terapia combinada con REVLIMID para MM y MCL
Dosis en la terapia combinada con REVLIMID para FL y LZM
Dosis en la terapia de mantenimiento con REVLIMID después del auto-HSCT para MM y para MDS
CLcr de 30 a 60 ml/min
10 mg una vez al día
10 mg una vez al día
5 mg una vez al día
CLcr inferior a 30 ml/min (que no requiere diálisis)
15 mg en días alternos
5 mg una vez al día
2,5 mg una vez al día
CLcr inferior a 30 ml/min (que requiere diálisis)
5 mg una vez al día. Los días de diálisis, administre la dosis después de la diálisis.
5 mg una vez al día. Los días de diálisis, administre la dosis después de la diálisis.
2,5 mg una vez al día. Los días de diálisis, administre la dosis después de la diálisis.
Terapia combinada con REVLIMID para el MM: Para una CLcr de 30 a 60 ml/min, considere aumentar la dosis a 15 mg después de 2 ciclos si el paciente tolera la dosis de 10 mg de lenalidomida sin toxicidad limitante de la dosis.
Terapia de mantenimiento con REVLIMID después de un auto-HSCT para MM y para MCL y MDS: Base el aumento o la disminución posterior de la dosis de REVLIMID en la tolerancia al tratamiento individual del paciente [ver Dosificación y administración (2.1– 2.3)].
Terapia combinada con REVLIMID para FL o para MZL: Para pacientes con una CLcr de 30 a 60 ml/min, después de 2 ciclos, la dosis de REVLIMID puede aumentarse a 15 mg por vía oral si el paciente ha tolerado la terapia.
2.7 Administración
Aconseje a los pacientes que tomen REVLIMID por vía oral aproximadamente a la misma hora todos los días, con o sin alimentos. Aconseje a los pacientes que traguen las cápsulas de REVLIMID enteras con agua y que no las abran, rompan ni mastiquen.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Cápsulas:
•
2.5 mg, cápsulas duras opacas blancas y azul verdosas impresas con “REV” en una mitad y “2.5 mg” en la otra mitad con tinta negra
•
5 mg, cápsulas opacas blancas impresas con “REV” en una mitad y “5 mg” en la otra mitad con tinta negra
•
10 mg, cápsulas opacas azul/verdes y amarillo pálido impresas con “REV” en una mitad y “10 mg” en la otra mitad con tinta negra
•
15 mg, cápsulas opacas azul claro y blancas impresas con “REV” en una mitad y “15 mg” en la otra mitad con tinta negra
•
20 mg, cápsulas duras opacas azul claro y azul verdosas impresas con “REV” en una mitad y “20 mg” en la otra mitad con tinta negra
•
25 mg, cápsulas opacas blancas impresas con “REV” en una mitad y “25 mg” en la otra mitad con tinta negra
4 CONTRAINDICACIONES
4.1 Embarazo
REVLIMID puede causar daño fetal cuando se administra a una mujer embarazada. Se observaron anormalidades en las extremidades en la descendencia de monos que recibieron lenalidomida durante la organogénesis. Este efecto se observó en todas las dosis probadas. Debido a los resultados de este estudio de desarrollo en monos y las similitudes estructurales de la lenalidomida con la talidomida, un teratógeno humano conocido, la lenalidomida está contraindicada en mujeres embarazadas [ver Advertencia en recuadro]. Si este medicamento se usa durante el embarazo o si la paciente queda embarazada mientras toma este medicamento, se le debe informar sobre el riesgo potencial para el feto [ver Advertencias y precauciones (5.1, 5.2), Uso en poblaciones especiales (8.1, 8.3)].
4.2 Reacciones de hipersensibilidad graves
REVLIMID está contraindicado en pacientes que han demostrado hipersensibilidad grave (por ejemplo, angioedema, síndrome de Stevens-Johnson, necrólisis epidérmica tóxica) a la lenalidomida [ver Advertencias y precauciones (5.9, 5.15)].
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Toxicidad embrio-fetal
REVLIMID es un análogo de la talidomida y está contraindicado para su uso durante el embarazo. La talidomida es un teratógeno humano conocido que causa defectos de nacimiento humanos que ponen en peligro la vida o muerte embrio-fetal [ver Uso en poblaciones específicas (8.1)]. Un estudio de desarrollo embrio-fetal en monos indica que la lenalidomida produjo malformaciones en la descendencia de monos hembra que recibieron el fármaco durante el embarazo, similar a los defectos de nacimiento observados en humanos después de la exposición a la talidomida durante el embarazo.
Las mujeres en edad fértil deben evitar el embarazo durante al menos 4 semanas antes de comenzar el tratamiento con REVLIMID, durante el tratamiento, durante las interrupciones de la dosis y durante al menos 4 semanas después de completar el tratamiento.
Las mujeres deben comprometerse a abstenerse continuamente de las relaciones sexuales heterosexuales o a utilizar dos métodos de control de la natalidad fiables, comenzando 4 semanas antes de iniciar el tratamiento con REVLIMID, durante el tratamiento, durante las interrupciones de la dosis y continuando durante 4 semanas después de la interrupción del tratamiento con REVLIMID.
Se deben obtener dos pruebas de embarazo negativas antes de iniciar el tratamiento. La primera prueba debe realizarse dentro de los 10-14 días y la segunda prueba dentro de las 24 horas previas a la prescripción del tratamiento con REVLIMID y luego semanalmente durante el primer mes, luego mensualmente a partir de entonces en mujeres con ciclos menstruales regulares o cada 2 semanas en mujeres con ciclos menstruales irregulares [ver Uso en poblaciones específicas (8.3)].
Hombres
La lenalidomida está presente en el semen de los pacientes que reciben el fármaco. Por lo tanto, los hombres deben usar siempre un condón de látex o sintético durante cualquier contacto sexual con mujeres en edad fértil mientras toman REVLIMID y hasta 4 semanas después de interrumpir REVLIMID, incluso si se han sometido a una vasectomía exitosa. Los pacientes masculinos que toman REVLIMID no deben donar esperma y hasta 4 semanas después de interrumpir REVLIMID [ver Uso en poblaciones específicas (8.3)].
Donación de sangre
Los pacientes no deben donar sangre durante el tratamiento con REVLIMID y durante 4 semanas después de la interrupción del fármaco porque la sangre podría administrarse a una paciente embarazada cuyo feto no debe estar expuesto a REVLIMID.
5.2 Programa Lenalidomide REMS
Debido al riesgo embrio-fetal [ver Advertencias y precauciones (5.1)], REVLIMID solo está disponible a través de un programa restringido bajo una Estrategia de Evaluación y Mitigación de Riesgos (REMS), el programa Lenalidomide REMS.
Los componentes necesarios del programa Lenalidomide REMS incluyen los siguientes:
•
Los prescriptores deben estar certificados con el programa Lenalidomide REMS inscribiéndose y cumpliendo con los requisitos de REMS.
•
Los pacientes deben firmar un formulario de acuerdo paciente-médico y cumplir con los requisitos de REMS. En particular, las pacientes en edad fértil que no estén embarazadas deben cumplir con los requisitos de pruebas de embarazo y anticoncepción [ver Uso en poblaciones específicas (8.3)] y los hombres deben cumplir con los requisitos de anticoncepción [ver Uso en poblaciones específicas (8.3)].
•
Las farmacias deben estar certificadas con el programa Lenalidomide REMS, solo deben dispensar a pacientes que estén autorizados a recibir REVLIMID y cumplir con los requisitos de REMS.
Se puede obtener más información sobre el programa Lenalidomide REMS en www.lenalidomiderems.com o por teléfono al 1-888-423-5436.
5.3 Toxicidad hematológica
REVLIMID puede causar neutropenia y trombocitopenia significativas. Monitorear a los pacientes con neutropenia para detectar signos de infección. Aconsejar a los pacientes que observen si hay sangrado o hematomas, especialmente con el uso de medicamentos concomitantes que pueden aumentar el riesgo de sangrado. Los pacientes que toman REVLIMID deben hacerse análisis de sangre completos periódicamente como se describe a continuación [ver Dosificación y administración (2.1, 2.2, 2.3)].
Monitorear los recuentos sanguíneos completos (CBC) en pacientes que toman REVLIMID en combinación con dexametasona o como terapia de mantenimiento con REVLIMID para MM cada 7 días (semanalmente) durante los primeros 2 ciclos, en los días 1 y 15 del ciclo 3, y cada 28 días (4 semanas) a partir de entonces. Puede ser necesaria una interrupción de la dosis y/o una reducción de la dosis [ver Dosificación y administración (2.1)]. En los ensayos de terapia de mantenimiento de MM, se informó neutropenia de grado 3 o 4 en hasta el 59% de los pacientes tratados con REVLIMID y trombocitopenia de grado 3 o 4 en hasta el 38% de los pacientes tratados con REVLIMID [ver Reacciones adversas (6.1)].
Controle los recuentos sanguíneos completos (CBC) en pacientes que toman REVLIMID para MDS semanalmente durante las primeras 8 semanas y al menos mensualmente a partir de entonces. La toxicidad hematológica de grado 3 o 4 se observó en el 80% de los pacientes inscritos en el estudio de MDS. En el 48% de los pacientes que desarrollaron neutropenia de grado 3 o 4, el tiempo medio hasta el inicio fue de 42 días (rango, 14-411 días), y el tiempo medio hasta la recuperación documentada fue de 17 días (rango, 2-170 días). En el 54% de los pacientes que desarrollaron trombocitopenia de grado 3 o 4, el tiempo medio hasta el inicio fue de 28 días (rango, 8-290 días), y el tiempo medio hasta la recuperación documentada fue de 22 días (rango, 5-224 días) [ver Advertencia en recuadro y Dosificación y administración (2.2)].
Controle los recuentos sanguíneos completos (CBC) en pacientes que toman REVLIMID para MCL semanalmente durante el primer ciclo (28 días), cada 2 semanas durante los ciclos 2-4, y luego mensualmente a partir de entonces. Los pacientes pueden requerir interrupción de la dosis y/o reducción de la dosis. En el ensayo de MCL, se informó neutropenia de grado 3 o 4 en el 43% de los pacientes. Se informó trombocitopenia de grado 3 o 4 en el 28% de los pacientes.
Controle los recuentos sanguíneos completos (CBC) en pacientes que toman REVLIMID para FL o MZL semanalmente durante las primeras 3 semanas del Ciclo 1 (28 días), cada 2 semanas durante los Ciclos 2-4, y luego mensualmente a partir de entonces. Los pacientes pueden requerir interrupción de la dosis y/o reducción de la dosis. En los ensayos AUGMENT y MAGNIFY, se informó neutropenia de grado 3 o 4 en el 50% y el 33%, respectivamente, de los pacientes en el brazo REVLIMID/rituximab. Se informó trombocitopenia de grado 3 o 4 en el 2% y el 8%, respectivamente, de los pacientes en el brazo REVLIMID/rituximab [ver Reacciones adversas (6.1)].
5.4 Tromboembolismo venoso y arterial
Los eventos tromboembólicos venosos (VTE [DVT y PE]) y los eventos tromboembólicos arteriales (ATE, infarto de miocardio y accidente cerebrovascular) aumentan en los pacientes tratados con REVLIMID.
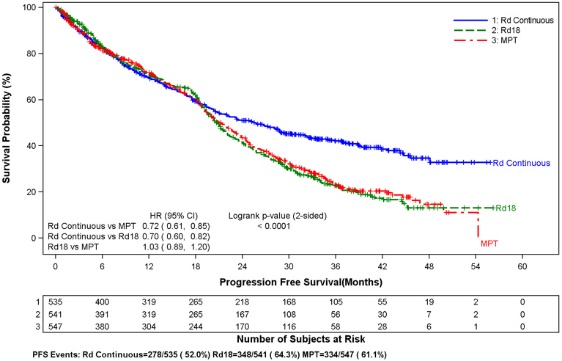
Se produjo un riesgo significativamente mayor de DVT (7,4%) y de PE (3,7%) en pacientes con MM después de al menos una terapia previa que fueron tratados con REVLIMID y terapia con dexametasona en comparación con los pacientes tratados en el grupo placebo y dexametasona (3,1% y 0,9%) en ensayos clínicos con uso variable de terapias anticoagulantes. En el estudio de mieloma múltiple de nuevo diagnóstico (NDMM) en el que casi todos los pacientes recibieron profilaxis antitrombótica, la DVT se informó como una reacción adversa grave (3,6%, 2,0% y 1,7%) en los brazos Rd Continuo, Rd18 y MPT, respectivamente. La frecuencia de reacciones adversas graves de PE fue similar entre los brazos Rd Continuo, Rd18 y MPT (3,8%, 2,8% y 3,7%, respectivamente) [ver Advertencia en recuadro y Reacciones adversas (6.1)].
El infarto de miocardio (1,7%) y el accidente cerebrovascular (CVA) (2,3%) aumentan en los pacientes con MM después de al menos una terapia previa que fueron tratados con REVLIMID y terapia con dexametasona en comparación con los pacientes tratados con placebo y dexametasona (0,6% y 0,9%) en ensayos clínicos. En el estudio de NDMM, el infarto de miocardio (incluido el agudo) se informó como una reacción adversa grave (2,3%, 0,6% y 1,1%) en los brazos Rd Continuo, Rd18 y MPT, respectivamente. La frecuencia de reacciones adversas graves de CVA fue similar entre los brazos Rd Continuo, Rd18 y MPT (0,8%, 0,6% y 0,6%, respectivamente) [ver Reacciones adversas (6.1)].
Los pacientes con factores de riesgo conocidos, incluida la trombosis previa, pueden tener un mayor riesgo y se deben tomar medidas para tratar de minimizar todos los factores modificables (por ejemplo, hiperlipidemia, hipertensión, tabaquismo).
En ensayos clínicos controlados que no utilizaron tromboprofilaxis concomitante, se produjeron eventos trombóticos generales del 21,5% (consulta estandarizada de MedDRA sobre eventos embólicos y trombóticos) en pacientes con MM refractario y recidivante que fueron tratados con REVLIMID y dexametasona en comparación con el 8,3% de trombosis en pacientes tratados con placebo y dexametasona. El tiempo medio hasta el primer evento trombótico fue de 2,8 meses. En el estudio de NDMM en el que casi todos los pacientes recibieron profilaxis antitrombótica, la frecuencia general de eventos trombóticos fue del 17,4% en los pacientes de los brazos combinados Rd Continuo y Rd18, y del 11,6% en el brazo MPT. El tiempo medio hasta el primer evento trombótico fue de 4,3 meses en los brazos combinados Rd Continuo y Rd18.
En el ensayo AUGMENT, la incidencia de VTE (incluidas DVT y PE) en pacientes con FL o MZL fue del 3,4% en el brazo REVLIMID/rituximab [ver Reacciones adversas (6.1)]. En el ensayo AUGMENT, la incidencia de ATE (incluido el IM) en pacientes con FL o MZL fue del 0,6% en el brazo REVLIMID/rituximab [ver Reacciones adversas (6.1)].
Se recomienda la tromboprofilaxis. El régimen de tromboprofilaxis debe basarse en una evaluación de los riesgos subyacentes del paciente. Indique a los pacientes que informen inmediatamente cualquier signo o síntoma que sugiera eventos trombóticos. Los ESA y los estrógenos pueden aumentar aún más el riesgo de trombosis y su uso debe basarse en una decisión de beneficio-riesgo en pacientes que reciben REVLIMID [ver Interacciones medicamentosas (7.2)].
5.5 Aumento de la mortalidad en pacientes con CLL
En un ensayo clínico prospectivo aleatorizado (1:1) en el tratamiento de primera línea de pacientes con leucemia linfocítica crónica, la terapia con REVLIMID en monoterapia aumentó el riesgo de muerte en comparación con la clorambucil en monoterapia. En un análisis intermedio, hubo 34 muertes entre 210 pacientes en el brazo de tratamiento con REVLIMID en comparación con 18 muertes entre 211 pacientes en el brazo de tratamiento con clorambucil, y la razón de riesgo para la supervivencia general fue de 1,92 [IC del 95%: 1,08 – 3,41], consistente con un aumento del 92% en el riesgo de muerte. El ensayo se detuvo por seguridad en julio de 2013.
Las reacciones adversas cardiovasculares graves, incluida la fibrilación auricular, el infarto de miocardio y la insuficiencia cardíaca, ocurrieron con mayor frecuencia en el brazo de tratamiento con REVLIMID. REVLIMID no está indicado ni recomendado para su uso en CLL fuera de ensayos clínicos controlados.
5.6 Segundas neoplasias malignas primarias
En ensayos clínicos en pacientes con MM que recibieron REVLIMID, se ha observado un aumento de las segundas neoplasias malignas primarias (SPM) hematológicas más tumores sólidos, en particular AML y MDS. Se produjo un aumento de las SPM hematológicas, incluidas AML y MDS, en el 5,3% de los pacientes con NDMM que recibieron REVLIMID en combinación con melfalán oral en comparación con el 1,3% de los pacientes que recibieron melfalán sin REVLIMID. La frecuencia de los casos de AML y MDS en pacientes con NDMM tratados con REVLIMID en combinación con dexametasona sin melfalán fue del 0,4%.
En pacientes que recibieron terapia de mantenimiento con REVLIMID después de melfalán intravenoso de alta dosis y auto-HSCT, SPM hematológico ocurrió en el 7.5% de los pacientes en comparación con el 3.3% en los pacientes que recibieron placebo. La incidencia de SPM hematológico más tumor sólido (excluyendo carcinoma de células escamosas y carcinoma de células basales) fue del 14.9%, en comparación con el 8.8% en los pacientes que recibieron placebo con un seguimiento mediano de 91.5 meses. El SPM de cáncer de piel no melanoma, incluido el carcinoma de células escamosas y el carcinoma de células basales, ocurrió en el 3.9% de los pacientes que recibieron mantenimiento con REVLIMID, en comparación con el 2.6% en el brazo placebo.
En pacientes con MM recidivante o refractario tratados con REVLIMID/dexametasona, la incidencia de SPM hematológico más tumor sólido (excluyendo carcinoma de células escamosas y carcinoma de células basales) fue del 2.3% versus 0.6% en el brazo de dexametasona sola. El SPM de cáncer de piel no melanoma, incluido el carcinoma de células escamosas y el carcinoma de células basales, ocurrió en el 3.1% de los pacientes que recibieron REVLIMID/dexametasona, en comparación con el 0.6% en el brazo de dexametasona sola.
Los pacientes que recibieron terapia con REVLIMID hasta la progresión de la enfermedad no mostraron una mayor incidencia de SPM invasivo que los pacientes tratados en los brazos con REVLIMID de duración fija. Monitorear a los pacientes para el desarrollo de segundas neoplasias malignas. Tener en cuenta tanto el beneficio potencial de REVLIMID como el riesgo de segundas neoplasias malignas al considerar el tratamiento con REVLIMID.
En el ensayo AUGMENT con pacientes con FL o MZL que recibieron terapia con REVLIMID/rituximab, se han observado SPM hematológicos más tumor sólido, en particular AML. En el ensayo AUGMENT, el SPM hematológico de AML ocurrió en el 0.6% de los pacientes con FL o MZL que recibieron terapia con REVLIMID/rituximab. La incidencia de SPM hematológico más tumor sólido (excluyendo cánceres de piel no melanoma) fue del 1.7% en el brazo de REVLIMID/rituximab con un seguimiento mediano de 29.8 meses (rango de 0.5 a 51.3 meses) [ver Reacciones adversas (6.1)]. Monitorear a los pacientes para el desarrollo de segundas neoplasias malignas. Tener en cuenta tanto el beneficio potencial de REVLIMID como el riesgo de segundas neoplasias malignas al considerar el tratamiento con REVLIMID.
5.7 Aumento de la mortalidad en pacientes con MM cuando se agrega pembrolizumab a un análogo de talidomida y dexametasona
En dos ensayos clínicos aleatorizados en pacientes con MM, la adición de pembrolizumab a un análogo de talidomida más dexametasona, un uso para el cual no está indicado ningún anticuerpo bloqueador de PD-1 o PD-L1, resultó en un aumento de la mortalidad. No se recomienda el tratamiento de pacientes con MM con un anticuerpo bloqueador de PD-1 o PD-L1 en combinación con un análogo de talidomida más dexametasona fuera de los ensayos clínicos controlados.
5.8 Hepatotoxicidad
Se ha producido insuficiencia hepática, incluidos casos mortales, en pacientes tratados con REVLIMID en combinación con dexametasona. En los ensayos clínicos, el 15% de los pacientes experimentaron hepatotoxicidad (con características hepatocelulares, colestásicas y mixtas); el 2% de los pacientes con MM y el 1% de los pacientes con mielodisplasia tuvieron eventos graves de hepatotoxicidad. El mecanismo de la hepatotoxicidad inducida por fármacos es desconocido. La enfermedad hepática viral preexistente, las enzimas hepáticas basales elevadas y los medicamentos concomitantes pueden ser factores de riesgo. Monitorear las enzimas hepáticas periódicamente. Detenga REVLIMID si las enzimas hepáticas se elevan. Después de volver a los valores basales, se puede considerar el tratamiento a una dosis más baja.
5.9 Reacciones cutáneas graves
Se han notificado reacciones cutáneas graves, incluido el síndrome de Stevens-Johnson (SSJ), la necrólisis epidérmica tóxica (NET) y la reacción medicamentosa con eosinofilia y síntomas sistémicos (DRESS). El DRESS puede presentarse con una reacción cutánea (como erupción o dermatitis exfoliativa), eosinofilia, fiebre y/o linfadenopatía con complicaciones sistémicas como hepatitis, nefritis, neumonitis, miocarditis y/o pericarditis. Estos eventos pueden ser fatales. Los pacientes con antecedentes de erupción de grado 4 asociada con el tratamiento con talidomida no deben recibir REVLIMID. Considere la interrupción o la interrupción de REVLIMID para la erupción cutánea de grado 2-3. Suspenda permanentemente REVLIMID para la erupción de grado 4, la erupción exfoliativa o bullosa, o para otras reacciones cutáneas graves como SSJ, NET o DRESS [ver Dosificación y administración (2.5)].
5.10 Síndrome de lisis tumoral
Se han notificado casos mortales de síndrome de lisis tumoral (SLT) durante el tratamiento con REVLIMID. Los pacientes con riesgo de SLT son aquellos con una alta carga tumoral antes del tratamiento. Monitorear a los pacientes en riesgo de cerca y tomar las medidas preventivas apropiadas. En el ensayo AUGMENT en pacientes con FL o MZL, el SLT ocurrió en 2 pacientes (1.1%) en el brazo de REVLIMID/rituximab. El SLT ocurrió en 1 paciente (0.5%) en el ensayo MAGNIFY durante el período de inducción de REVLIMID/rituximab; el evento fue una reacción adversa grave de grado 3.
5.11 Reacción de brote tumoral
La reacción de brote tumoral (RBT), incluidas las reacciones fatales, se ha producido durante el uso de investigación de REVLIMID para la LLC y el linfoma, y se caracteriza por hinchazón de los ganglios linfáticos sensibles, fiebre de bajo grado, dolor y erupción. REVLIMID no está indicado ni recomendado para su uso en la LLC fuera de los ensayos clínicos controlados.
Se recomienda el monitoreo y la evaluación de la RBT en pacientes con MCL, FL o MZL. La reacción de brote tumoral puede imitar la progresión de la enfermedad (PD).
En el ensayo MCL, 13/134 (10%) de los sujetos experimentaron RBT; todos los informes fueron de grado 1 o 2 en gravedad. Todos los eventos ocurrieron en el ciclo 1 y un paciente desarrolló RBT nuevamente en el ciclo 11. En el ensayo AUGMENT en pacientes con FL o MZL, se informó RBT en 19/176 (10.8%) de los pacientes en el brazo de REVLIMID con rituximab; un paciente en el brazo de REVLIMID/rituximab experimentó una RBT de grado 3. En el ensayo MAGNIFY, 9/222 (4.1%) de los pacientes experimentaron RBT; todos los informes fueron de grado 1 o 2 en gravedad y 1 evento se consideró grave. En un ensayo de fase 2 separado de MCL, un caso de RBT resultó en un resultado fatal.
REVLIMID puede continuarse en pacientes con RBT de grado 1 y 2 sin interrupción o modificación, a discreción del médico. Los pacientes con RBT de grado 1 y 2 también pueden ser tratados con corticosteroides, medicamentos antiinflamatorios no esteroideos (AINE) y/o analgésicos narcóticos para el manejo de los síntomas de la RBT. En pacientes con RBT de grado 3 o 4, se recomienda suspender el tratamiento con REVLIMID hasta que la RBT se resuelva a ≤ grado 1. Los pacientes con RBT de grado 3 o 4 pueden ser tratados para el manejo de los síntomas según la guía para el tratamiento de la RBT de grado 1 y 2.
5.12 Movilización de Células Madre Deteriorada
Se ha informado una disminución en el número de células CD34+ recolectadas después del tratamiento (> 4 ciclos) con REVLIMID. En pacientes que son candidatos a auto-HSCT, la derivación a un centro de trasplante debe ocurrir temprano en el tratamiento para optimizar el momento de la recolección de células madre. En pacientes que recibieron más de 4 ciclos de un tratamiento que contiene REVLIMID o para quienes no se han recolectado cantidades adecuadas de células CD 34+ con G-CSF solo, se puede considerar G-CSF con ciclofosfamida o la combinación de G-CSF con un inhibidor de CXCR4.
5.13 Trastornos de la Tiroides
Se han informado tanto hipotiroidismo como hipertiroidismo [ver Reacciones adversas (6.2)]. Mida la función tiroidea antes de comenzar el tratamiento con REVLIMID y durante la terapia.
5.14 Mortalidad Temprana en Pacientes con MCL
En otro estudio de MCL, hubo un aumento en las muertes tempranas (dentro de las 20 semanas), 12.9% en el brazo de REVLIMID versus 7.1% en el brazo de control. En el análisis multivariado exploratorio, los factores de riesgo para las muertes tempranas incluyen una alta carga tumoral, la puntuación MIPI en el diagnóstico y un alto recuento de glóbulos blancos al inicio (≥ 10 x 109/L).
5.15 Hipersensibilidad
Se ha informado hipersensibilidad, incluido angioedema, anafilaxia y reacciones anafilácticas a REVLIMID. Suspenda permanentemente REVLIMID para angioedema y anafilaxia [ver Dosificación y administración (2.2)].
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas clínicamente significativas se describen en detalle en otras secciones de la información de prescripción:
Mortalidad aumentada en pacientes con MM cuando Pembrolizumab se añade a un análogo de talidomida y dexametasona [véase Advertencias y Precauciones (5.7)]
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variadas, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y puede que no reflejen las tasas observadas en la práctica.
MM recién diagnosticado – Terapia combinada con REVLIMID:
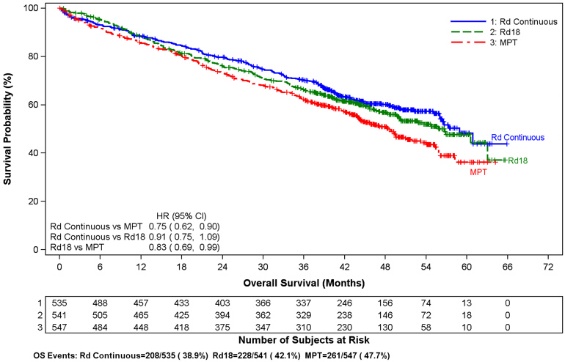
Se evaluaron los datos de 1613 pacientes en un estudio de fase 3 extenso que recibieron al menos una dosis de REVLIMID con dexametasona en baja dosis (Rd) durante 2 duraciones de tiempo diferentes (es decir, hasta la enfermedad progresiva [Brazo Rd Continuo; N = 532] o durante un máximo de dieciocho ciclos de 28 días [72 semanas, Brazo Rd18; N = 540] o que recibieron melphalan, prednisona y talidomida (Brazo MPT; N = 541) durante un máximo de doce ciclos de 42 días (72 semanas). La duración mediana del tratamiento en el brazo Rd Continuo fue de 80,2 semanas (rango 0,7 a 246,7) o 18,4 meses (rango 0,16 a 56,7).
En general, las reacciones adversas más frecuentemente notificadas fueron comparables en el brazo Rd Continuo y el brazo Rd18 e incluyeron diarrea, anemia, estreñimiento, edema periférico, neutropenia, fatiga, dolor de espalda, náuseas, astenia e insomnio. Las reacciones de Grado 3 o 4 más frecuentemente reportadas incluyeron neutropenia, anemia, trombocitopenia, neumonía, astenia, fatiga, dolor de espalda, hipopotasemia, erupción cutánea, catarata, linfopenia, disnea, TEP, hiperglucemia y leucopenia. La mayor frecuencia de infecciones ocurrió en el brazo Rd Continuo (75%) en comparación con el brazo MPT (56%). Hubo más reacciones adversas de infección de Grado 3 y 4 y graves en el brazo Rd Continuo que en el brazo MPT o el Rd18.
En el brazo Rd Continuo, las reacciones adversas más comunes que llevaron a la interrupción de la dosis de REVLIMID fueron eventos infecciosos (28,8%); en general, el tiempo mediano hasta la primera interrupción de la dosis de REVLIMID fue de 7 semanas. Las reacciones adversas más comunes que llevaron a la reducción de la dosis de REVLIMID en el brazo Rd Continuo fueron eventos hematológicos (10,7%); en general, el tiempo mediano hasta la primera reducción de la dosis de REVLIMID fue de 16 semanas. En el brazo Rd Continuo, las reacciones adversas más comunes que llevaron a la discontinuación de REVLIMID fueron eventos infecciosos (3,4%).
En ambos brazos Rd, las frecuencias de inicio de las reacciones adversas fueron generalmente más altas durante los primeros 6 meses del tratamiento y luego las frecuencias disminuyeron con el tiempo o se mantuvieron estables durante todo el tratamiento, excepto para las cataratas. La frecuencia de inicio de las cataratas aumentó con el tiempo con 0,7% durante los primeros 6 meses y hasta 9,6% en el 2º año de tratamiento con Rd Continuo.
La Tabla 4 resume las reacciones adversas notificadas para los brazos de tratamiento Rd Continuo, Rd18 y MPT.
Tabla 4: Todas las reacciones adversas en ≥5% y reacciones adversas de grado 3/4 en ≥1% de los pacientes con MM en los brazos Rd Continuo o Rd18*
Nota: Un sujeto con múltiples ocurrencias de una reacción adversa se cuenta solo una vez bajo el Sistema Corporal/Reacción Adversa aplicable. a Todos los eventos adversos emergentes del tratamiento en al menos el 5% de los sujetos en los brazos Rd Continuo o Rd18 y al menos una frecuencia (%) 2% más alta en cualquiera de los brazos Rd Continuo o Rd18 en comparación con el brazo MPT. b Todos los eventos adversos emergentes del tratamiento de grado 3 o 4 en al menos el 1% de los sujetos en los brazos Rd Continuo o Rd18 y al menos una frecuencia (%) 1% más alta en cualquiera de los brazos Rd Continuo o Rd18 en comparación con el brazo MPT. c Eventos adversos emergentes del tratamiento graves en al menos el 1% de los sujetos en los brazos Rd Continuo o Rd18 y al menos una frecuencia (%) 1% más alta en cualquiera de los brazos Rd Continuo o Rd18 en comparación con el brazo MPT. d Los términos preferidos para el sistema corporal de trastornos de la sangre y el sistema linfático se incluyeron por juicio médico como reacciones adversas conocidas para Rd Continuo/Rd18, y también se han reportado como graves. e Pie de página “a” no aplicable. f Pie de página “b” no aplicable. @ – reacciones adversas en las que al menos una resultó en un desenlace fatal. % – reacciones adversas en las que al menos una se consideró que ponía en peligro la vida (si el desenlace de la reacción fue la muerte, se incluye con los casos de muerte). *Reacciones adversas incluidas en términos combinados de reacciones adversas: Dolor abdominal: Dolor abdominal, dolor abdominal superior, dolor abdominal inferior, dolor gastrointestinal Neumonías: Neumonía, neumonía lobar, neumonía neumocócica, bronconeumonía, neumonía por Pneumocystis jiroveci, neumonía por legionella, neumonía estafilocócica, neumonía por klebsiella, neumonía atípica, neumonía bacteriana, neumonía por escherichia, neumonía estreptocócica, neumonía viral Sepsis: Sepsis, shock séptico, urosepsis, sepsis por escherichia, sepsis neutropénica, sepsis neumocócica, sepsis estafilocócica, sepsis bacteriana, sepsis meningocócica, sepsis enterococo, sepsis por klebsiella, sepsis pseudomonal Erupción: Erupción, erupción pruriginosa, erupción eritematosa, erupción maculo-papular, erupción generalizada, erupción papular, erupción exfoliativa, erupción folicular, erupción macular, erupción medicamentosa con eosinofilia y síntomas sistémicos, eritema multiforme, erupción pustular Trombosis venosa profunda: Trombosis venosa profunda, trombosis venosa de extremidades, trombosis venosa
Sistema corporal
Reacción adversa
Todas las reacciones adversasa
Reacciones adversas de grado 3/4b
Rd Continuo
(N = 532)
Rd18
(N = 540)
MPT
(N = 541)
Rd Continuo
(N = 532)
Rd18
(N = 540)
MPT
(N = 541)
Trastornos generales y condiciones del sitio de administración
Fatiga%
173 (33)
177 (33)
154 (28)
39 ( 7)
46 ( 9)
31 ( 6)
Astenia
150 (28)
123 (23)
124 (23)
41 ( 8)
33 ( 6)
32 ( 6)
Pirexiac
114 (21)
102 (19)
76 (14)
13 ( 2)
7 ( 1)
7 ( 1)
Dolor torácico no cardíaco f
29 ( 5)
31 ( 6)
18 ( 3)
<1%
< 1%
< 1%
Trastornos gastrointestinales
Diarrea
242 (45)
208 (39)
89 (16)
21 ( 4)
18 ( 3)
8 ( 1)
Dolor abdominal% f
109 (20)
78 (14)
60 (11)
7 ( 1)
9 ( 2)
< 1%
Dispepsia f
57 (11)
28 ( 5)
36 ( 7)
<1%
< 1%
0 ( 0)
Trastornos musculoesqueléticos y del tejido conjuntivo
Dolor de espaldac
170 (32)
145 (27)
116 (21)
37 ( 7)
34 ( 6)
28 ( 5)
Espasmos musculares f
109 (20)
102 (19)
61 (11)
< 1%
< 1%
< 1%
Artralgia f
101 (19)
71 (13)
66 (12)
9 ( 2)
8 ( 1)
8 ( 1)
Dolor óseo f
87 (16)
77 (14)
62 (11)
16 ( 3)
15 ( 3)
14 ( 3)
Dolor en las extremidades f
79 (15)
66 (12)
61 (11)
8 ( 2)
8 ( 1)
7 ( 1)
Dolor musculoesquelético f
67 (13)
59 (11)
36 ( 7)
< 1%
< 1%
< 1%
Dolor torácico musculoesquelético f
60 (11)
51 ( 9)
39 ( 7)
6 ( 1)
< 1%
< 1%
Debilidad muscular f
43 ( 8)
35 ( 6)
29 ( 5)
< 1%
8 ( 1)
< 1%
Dolor de cuello f
40 ( 8)
19 ( 4)
10 ( 2)
< 1%
< 1%
< 1%
Infecciones e infestaciones
Bronquitisc
90 (17)
59 (11)
43 ( 8)
9 ( 2)
6 ( 1)
< 1%
Nasofaringitis f
80 (15)
54 (10)
33 ( 6)
0 ( 0)
0 ( 0)
0 ( 0)
Infección del tracto urinario f
76 (14)
63 (12)
41 ( 8)
8 ( 2)
8 ( 1)
< 1%
Infección de las vías respiratorias superioresc% f
69 (13)
53 ( 10)
31 ( 6)
< 1%
8 ( 1)
< 1%
Neumoníac@
93 (17)
87 (16)
56 (10)
60 (11)
57 (11)
41 ( 8)
Infección del tracto respiratorio%
35 ( 7)
25 ( 5)
21 ( 4)
7 ( 1)
< 1%
< 1%
Influenza f
33 ( 6)
23 ( 4)
15 ( 3)
< 1%
< 1%
0 ( 0)
Gastroenteritis f
32 ( 6)
17 ( 3)
13 ( 2)
0 ( 0)
< 1%
< 1%
Infección del tracto respiratorio inferior
29 ( 5)
14 ( 3)
16 ( 3)
10 ( 2)
< 1%
< 1%
Rinitis f
29 ( 5)
24 ( 4)
14 ( 3)
0 ( 0)
0 ( 0)
0 ( 0)
Celulitisc
< 5%
< 5%
< 5%
8 ( 2)
< 1%
< 1%
Sepsisc@
33 ( 6)
26 ( 5)
18 ( 3)
26 ( 5)
20 ( 4)
13 ( 2)
Trastornos del sistema nervioso
Dolor de cabeza f
75 (14)
52 ( 10)
56 (10)
< 1%
< 1%
< 1%
Disgeusia f
39 ( 7)
45 ( 8)
22 ( 4)
< 1%
0 ( 0.0)
< 1%
Trastornos de la sangre y del sistema linfáticod
Anemia
233 (44)
193 (36)
229 (42)
97 (18)
85 (16)
102 (19)
Neutropenia
186 (35)
178 (33)
328 (61)
148 (28)
143 (26)
243 (45)
Trombocitopenia
104 (20)
100 (19)
135 (25)
44 ( 8)
43 ( 8)
60 (11)
Neutropenia febril
7 ( 1)
17 ( 3)
15 ( 3)
6 ( 1)
16 ( 3)
14 ( 3)
Pancytopenia
< 1%
6 ( 1)
7 ( 1)
< 1%
< 1%
< 1%
Trastornos respiratorios, torácicos y mediastínicos
Tos f
121 (23)
94 (17)
68 (13)
< 1%
< 1%
< 1%
Disneac,e
117 (22)
89 (16)
113 (21)
30 ( 6)
22 ( 4)
18 ( 3)
Epistaxis f
32 ( 6)
31 ( 6)
17 ( 3)
< 1%
< 1%
0 ( 0)
Dolor orofaríngeo f
30 ( 6)
22 ( 4)
14 ( 3)
0 ( 0)
0 ( 0)
0 ( 0)
Disnea de esfuerzo e
27 ( 5)
29 ( 5)
< 5%
6 ( 1)
< 1%
0 ( 0)
Trastornos del metabolismo y la nutrición
Disminución del apetito
123 (23)
115 (21)
72 (13)
14 ( 3)
7 ( 1)
< 1%
Hipokalemia%
91 (17)
62 (11)
38 ( 7)
35 ( 7)
20 ( 4)
11 ( 2)
Hiperglucemia
62 (12)
52 (10)
19 ( 4)
28 ( 5)
23 ( 4)
9 ( 2)
Hipocalcemia
57 (11)
56 (10)
31 ( 6)
23 ( 4)
19 ( 4)
8 ( 1)
Deshidratación%
25 ( 5)
29 ( 5)
17 ( 3)
8 ( 2)
13 ( 2)
9 ( 2)
Gota e
< 5%
< 5%
< 5%
8 ( 2)
0 ( 0)
0 ( 0)
Diabetes mellitus% e
< 5%
< 5%
< 5%
8 ( 2)
< 1%
< 1%
Hipo fosfatemia e
< 5%
< 5%
< 5%
7 ( 1)
< 1%
< 1%
Hiponatremia% e
< 5%
< 5%
< 5%
7 ( 1)
13 ( 2)
6 ( 1)
Trastornos de la piel y del tejido subcutáneo
Erupción
139 (26)
151 (28)
105 (19)
39 ( 7)
38 ( 7)
33 ( 6)
Prurito f
47 ( 9)
49 ( 9)
24 ( 4)
< 1%
< 1%
< 1%
Trastornos psiquiátricos
Insomnio
147 (28)
127 (24)
53 ( 10)
< 1%
6 ( 1)
0 ( 0)
Depresión
58 (11)
46 ( 9)
30 ( 6)
10 ( 2)
< 1%
< 1%
Trastornos vasculares
Trombosis venosa profundac%
55 (10)
39 ( 7)
22 ( 4)
30 ( 6)
20 ( 4)
15 ( 3)
Hipotensiónc%
51 (10)
35 ( 6)
36 ( 7)
11 ( 2)
8 ( 1)
6 ( 1)
Lesión, envenenamiento y complicaciones de procedimientos
Caída f
43 ( 8)
25 ( 5)
25 ( 5)
< 1%
6 ( 1)
6 ( 1)
Contusión f
33 ( 6)
24 ( 4)
15 ( 3)
< 1%
< 1%
0 ( 0)
Trastornos oculares
Catarata
73 (14)
31 ( 6)
< 1%
31 ( 6)
14 ( 3)
< 1%
Catarata subcapsular e
< 5%
< 5%
< 5%
7 ( 1)
0 ( 0)
0 ( 0)
Investigaciones
Pérdida de peso
72 (14)
78 (14)
48 ( 9)
11 ( 2)
< 1%
< 1%
Trastornos cardíacos
Fibrilación auricularc
37 ( 7)
25 ( 5)
25 ( 5)
13 ( 2)
9 ( 2)
6 ( 1)
Infarto de miocardio (incluido agudo)c ,e
< 5%
< 5%
< 5%
10 ( 2)
< 1%
< 1%
Trastornos renales y urinarios
Insuficiencia renal (incluida la aguda)c@,f
49 ( 9)
54 (10)
37 ( 7)
28 ( 5)
33 ( 6)
29 ( 5)
Neoplasias benignas, malignas e indeterminadas (incluidos quistes y pólipos)
Carcinoma de células escamosasc e
< 5%
< 5%
< 5%
8 ( 2)
< 1%
0 ( 0)
Carcinoma de células basalesc e,f
< 5%
< 5%
< 5%
< 1%
< 1%
0 ( 0)
MM de nuevo diagnóstico – Terapia de mantenimiento con REVLIMID después de auto-HSCT:
Se evaluaron datos de 1018 pacientes en dos ensayos aleatorizados que recibieron al menos una dosis de REVLIMID 10 mg diarios como terapia de mantenimiento después de auto-HSCT hasta la enfermedad progresiva o toxicidad inaceptable. La duración media del tratamiento con REVLIMID fue de 30,3 meses para el Estudio de mantenimiento 1 y 24,0 meses para el Estudio de mantenimiento 2 (rango general en ambos estudios de 0,1 a 108 meses). A la fecha de corte del 1 de marzo de 2015, 48 pacientes (21%) en el brazo de REVLIMID del Estudio de mantenimiento 1 todavía estaban en tratamiento y ninguno de los pacientes en el brazo de REVLIMID del Estudio de mantenimiento 2 todavía estaba en tratamiento en la misma fecha de corte.
Las reacciones adversas enumeradas del Estudio de mantenimiento 1 incluyeron eventos reportados después del trasplante (finalización de melfalán de alta dosis / auto-HSCT) y el período de tratamiento de mantenimiento. En el Estudio de mantenimiento 2, las reacciones adversas fueron solo del período de tratamiento de mantenimiento. En general, las reacciones adversas más frecuentes (más del 20% en el brazo de REVLIMID) en ambos estudios fueron neutropenia, trombocitopenia, leucopenia, anemia, infección del tracto respiratorio superior, bronquitis, nasofaringitis, tos, gastroenteritis, diarrea, erupción cutánea, fatiga, astenia, espasmo muscular y pirexia. Las reacciones de Grado 3 o 4 más frecuentes (más del 20% en el brazo de REVLIMID) incluyeron neutropenia, trombocitopenia y leucopenia. Las reacciones adversas graves infección pulmonar y neutropenia (más del 4,5%) ocurrieron en el brazo de REVLIMID.
Para REVLIMID, las reacciones adversas más comunes que llevaron a la interrupción de la dosis fueron eventos hematológicos (29,7%, datos disponibles solo en el Estudio de mantenimiento 2). La reacción adversa más común que llevó a la reducción de la dosis de REVLIMID fueron eventos hematológicos (17,7%, datos disponibles solo en el Estudio de mantenimiento 2). Las reacciones adversas más comunes que llevaron a la interrupción de REVLIMID fueron trombocitopenia (2,7%) en el Estudio de mantenimiento 1 y neutropenia (2,4%) en el Estudio de mantenimiento 2.
Las frecuencias de inicio de las reacciones adversas fueron generalmente más altas en los primeros 6 meses de tratamiento y luego las frecuencias disminuyeron con el tiempo o se mantuvieron estables durante todo el tratamiento.
La Tabla 5 resume las reacciones adversas reportadas para los brazos de tratamiento de mantenimiento con REVLIMID y placebo.
Tabla 5: Todas las reacciones adversas en ≥5% y reacciones adversas de Grado 3/4 en ≥1% de los pacientes con MM en los brazos de REVLIMID Vs Placebo*
Nota: Los eventos adversos (EA) se codifican en Sistema corporal / Reacción adversa utilizando MedDRA v15.1. Un sujeto con múltiples ocurrencias de una reacción adversa se cuenta solo una vez bajo el Sistema corporal / Reacción adversa aplicable. a Todos los EA emergentes del tratamiento en al menos el 5% de los pacientes en el grupo de mantenimiento de REVLIMID y al menos 2% de frecuencia (%) más alta que el grupo de mantenimiento de placebo. b Todos los EA emergentes del tratamiento de grado 3 o 4 en al menos el 1% de los pacientes en el grupo de mantenimiento de REVLIMID y al menos 1% de frecuencia (%) más alta que el grupo de mantenimiento de placebo. c Todos los EA emergentes del tratamiento graves en al menos el 1% de los pacientes en el grupo de mantenimiento de REVLIMID y al menos 1% de frecuencia (%) más alta que el grupo de mantenimiento de placebo. d La nota “a” no es aplicable para ninguno de los estudios e La nota “b” no es aplicable para ninguno de los estudios @ – ADR donde al menos uno resultó en un resultado fatal % – ADR donde al menos uno se consideró que ponía en peligro la vida (si el resultado del evento fue la muerte, se incluye con los casos de muerte) # – Todas las reacciones adversas bajo el Sistema corporal de Infecciones e Infestaciones, excepto las infecciones raras de interés para la salud pública, se considerarán enumeradas *Reacciones adversas para términos ADR combinados (basados en PT TEAE relevantes incluidos en los Estudios de mantenimiento 1 y 2 [según MedDRA v 15.1]): Neumonías Bronconeumonía, Neumonía lobar, Neumonía por Pneumocystis jiroveci, Neumonía, Neumonía klebsiella, Neumonía legionella, Neumonía micoplasmal, Neumonía neumocócica, Neumonía estreptocócica, Neumonía viral, Trastorno pulmonar, Neumonitis Sepsis: Sepsis bacteriana, Sepsis neumocócica, Sepsis, Shock séptico, Sepsis estafilocócica Neuropatía periférica: Neuropatía periférica, Neuropatía motora periférica, Neuropatía sensorial periférica, Polineuropatía Trombosis venosa profunda: Trombosis venosa profunda, Trombosis, Trombosis venosa
Sistema corporal
Reacción adversa
Estudio de mantenimiento 1
Estudio de mantenimiento 2
Todas las reacciones adversas a
Reacciones adversas de Grado 3/4 b
Todas las reacciones adversas a
Reacciones adversas de Grado 3/4 b
REVLIMID
(N=224)
n (%)
Placebo
(N=221)
n (%)
REVLIMID
(N=224)
n (%)
Placebo
(N=221)
n (%)
REVLIMID
(N=293)
n (%)
Placebo
(N=280)
n (%)
REVLIMID
(N=293)
n (%)
Placebo
(N=280)
n (%)
Trastornos de la sangre y del sistema linfático
Neutropenia c %
177 ( 79)
94 ( 43)
133 ( 59)
73 ( 33)
178 ( 61)
33 ( 12)
158 ( 54)
21 ( 8)
Trombocitopenia c %
162 ( 72)
101 ( 46)
84 ( 38)
67 ( 30)
69 ( 24)
29 ( 10)
38 ( 13)
8 ( 3)
Leucopenia c
51 ( 23)
25 ( 11)
45 ( 20)
22 ( 10)
93 ( 32)
21 ( 8)
71 ( 24)
5 ( 2)
Anemia
47 ( 21)
27 ( 12)
23 ( 10)
18 ( 8)
26 ( 9)
15 ( 5)
11 ( 4)
3 ( 1)
Linfopenia
40 ( 18)
29 ( 13)
37 ( 17)
26 ( 12)
13 ( 4)
3 ( 1)
11 ( 4)
< 1%
Pancytopenia c d %
< 1%
0 ( 0)
0 ( 0)
0 ( 0)
12 ( 4)
< 1%
7 ( 2)
< 1%
Neutropenia febril c
39 ( 17)
34 ( 15)
39 ( 17)
34 ( 15)
7 ( 2)
< 1%
5 ( 2)
< 1%
Infecciones e infestaciones#
Infección de las vías respiratorias altas e
60 ( 27)
35 ( 16)
7 ( 3)
9 ( 4)
32 ( 11)
18 ( 6)
< 1%
0 ( 0)
Infección neutropénica
40 ( 18)
19 ( 9)
27 ( 12)
14 ( 6)
0 ( 0)
0 ( 0)
0 ( 0)
0 ( 0)
Neumonías* c %
31 ( 14)
15 ( 7)
23 ( 10)
7 ( 3)
50 ( 17)
13 ( 5)
27 ( 9)
5 ( 2)
Bronquitis c
10 ( 4)
9 ( 4)
< 1%
5 ( 2)
139 ( 47)
104 ( 37)
4 ( 1)
< 1%
Nasofaringitis e
5 ( 2)
< 1%
0 ( 0)
0 ( 0)
102 ( 35)
84 ( 30)
< 1%
0 ( 0)
Gastroenteritis c
0 ( 0)
0 ( 0)
0 ( 0)
0 ( 0)
66 ( 23)
55 ( 20)
6 ( 2)
0 ( 0)
Rinitis e
< 1%
0 ( 0)
0 ( 0)
0 ( 0)
44 ( 15)
19 ( 7)
0 ( 0)
0 ( 0)
Sinusitis e
8 ( 4)
3 ( 1)
0 ( 0)
0 ( 0)
41 ( 14)
26 ( 9)
0 ( 0)
< 1%
Influenza c
8 ( 4)
5 ( 2)
< 1%
< 1%
39 ( 13)
19 ( 7)
3 ( 1)
0 ( 0)
Infección pulmonar c
21 ( 9)
< 1%
19 ( 8)
< 1%
9 ( 3)
4 ( 1)
< 1%
0 ( 0)
Infección del tracto respiratorio inferior e
13 ( 6)
5 ( 2)
6 ( 3)
4 ( 2)
4 ( 1)
4 ( 1)
0 ( 0)
< 1%
Infección c
12 ( 5)
6 ( 3)
9 ( 4)
5 ( 2)
17 ( 6)
5 ( 2)
0 ( 0)
0 ( 0)
Infección del tracto urinario c d e
9 ( 4)
5 ( 2)
4 ( 2)
4 ( 2)
22 ( 8)
17 ( 6)
< 1%
0 ( 0)
Infección del tracto respiratorio inferior bacteriana d
6 ( 3)
< 1%
4 ( 2)
0 ( 0)
0 ( 0)
0 ( 0)
0 ( 0)
0 ( 0)
Bacteriemia d
5 ( 2)
0 ( 0)
4 ( 2)
0 ( 0)
0 ( 0)
0 ( 0)
0 ( 0)
0 ( 0)
Herpes zóster c d
11 ( 5)
10 ( 5)
3 ( 1)
< 1%
29 ( 10)
25 ( 9)
6 ( 2)
< 1%
Sepsis* c d @
< 1%
< 1%
0 ( 0)
0 ( 0)
6 ( 2)
< 1%
4 ( 1)
< 1%
Trastornos gastrointestinales
Diarrea
122 ( 54)
83 ( 38)
22 ( 10)
17 ( 8)
114 ( 39)
34 ( 12)
7 ( 2)
0 ( 0)
Náuseas e
33 ( 15)
22 ( 10)
16 ( 7)
10 ( 5)
31 ( 11)
28 ( 10)
0 ( 0)
0 ( 0)
Vómitos
17 ( 8)
12 ( 5)
8 ( 4)
5 ( 2)
16 ( 5)
15 ( 5)
< 1%
0 ( 0)
Estreñimiento e
12 ( 5)
8 ( 4)
0 ( 0)
0 ( 0)
37 ( 13)
25 ( 9)
< 1%
0 ( 0)
Dolor abdominal e
8 ( 4)
7 ( 3)
< 1%
4 ( 2)
31 ( 11)
15 ( 5)
< 1%
< 1%
Dolor abdominal superior e
0 ( 0)
0 ( 0)
0 ( 0)
0 ( 0)
20 ( 7)
12 ( 4)
< 1%
0 ( 0)
Trastornos generales y condiciones del lugar de administración
Astenia
0 ( 0)
< 1%
0 ( 0)
0 ( 0)
87 ( 30)
53 ( 19)
10 ( 3)
< 1%
Fatiga
51 ( 23)
30 ( 14)
21 ( 9)
9 ( 4)
31 ( 11)
15 ( 5)
3 ( 1)
0 ( 0)
Pirexia e
17 ( 8)
10 ( 5)
< 1%
< 1%
60 ( 20)
26 ( 9)
< 1%
0 ( 0)
Trastornos de la piel y del tejido subcutáneo
Piel seca e
9 ( 4)
4 ( 2)
0 ( 0)
0 ( 0)
31 ( 11)
21 ( 8)
0 ( 0)
0 ( 0)
Erupción
71 ( 32)
48 ( 22)
11 ( 5)
5 ( 2)
22 ( 8)
17 ( 6)
3 ( 1)
0 ( 0)
Prurito
9 ( 4)
4 ( 2)
3 ( 1)
0 ( 0)
21 ( 7)
25 ( 9)
< 1%
0 ( 0)
Trastornos del sistema nervioso
Parestesia e
< 1%
0 ( 0)
0 ( 0)
0 ( 0)
39 ( 13)
30 ( 11)
< 1%
0 ( 0)
Neuropatía periférica* e
34 ( 15)
30 ( 14)
8 ( 4)
8 ( 4)
29 ( 10)
15 ( 5)
4 ( 1)
< 1%
Dolor de cabeza d
11 ( 5)
8 ( 4)
5 ( 2)
< 1%
25 ( 9)
21 ( 8)
0 ( 0)
0 ( 0)
Investigaciones
Alanina aminotransferasa aumentada
16 ( 7)
3 ( 1)
8 ( 4)
0 ( 0)
5 ( 2)
5 ( 2)
0 ( 0)
< 1%
Aspartato aminotransferasa aumentada d
13 ( 6)
5 ( 2)
6 ( 3)
0 ( 0)
< 1%
5 ( 2)
0 ( 0)
0 ( 0)
Trastornos del metabolismo y la nutrición
Hipokalemia
24 ( 11)
13 ( 6)
16 ( 7)
12 ( 5)
12 ( 4)
< 1%
< 1%
0 ( 0)
Deshidratación
9 ( 4 )
5 ( 2)
7 ( 3)
3 ( 1)
0 ( 0)
0 ( 0)
0 ( 0)
0 ( 0)
Hipo fosfatemia d
16 ( 7)
15 ( 7)
13 ( 6)
14 ( 6)
0 ( 0)
< 1%
0 ( 0)
0 ( 0)
Trastornos músculo esqueléticos y del tejido conjuntivo
Espasmos musculares e
0 ( 0)
< 1%
0 ( 0)
0 ( 0)
98 ( 33)
43 ( 15)
< 1%
0 ( 0)
Mialgia e
7 ( 3)
8 ( 4)
3 ( 1)
5 ( 2)
19 ( 6)
12 ( 4)
< 1%
< 1%
Dolor musculoesquelético e
< 1%
< 1%
0 ( 0)
0 ( 0)
19 ( 6)
11 ( 44)
0 ( 0)
0 ( 0)
Trastornos hepatobiliares
Hiperbilirrubinemia e
34 ( 15)
19 ( 9)
4 ( 2)
< 1%
4 ( 1)
< 1%
< 1%
0 ( 0)
Trastornos respiratorios, torácicos y mediastínicos
Tos e
23 ( 10)
12 ( 5)
3 ( 1)
< 1%
80 ( 27)
56 ( 20)
0 ( 0)
0 ( 0)
Disnea c e
15 ( 7)
9 ( 4)
8 ( 4)
4 ( 2)
17 ( 6)
9 ( 3)
< 1%
0 ( 0)
Rinorrea e
0 ( 0)
3 ( 1)
0 ( 0)
0 ( 0)
15 ( 5)
6 ( 2)
0 ( 0)
0 ( 0)
Embolia pulmonar c d e
0 ( 0)
0 ( 0)
0 ( 0)
0 ( 0)
3 ( 1)
0 ( 0)
< 1%
0 ( 0)
Trastornos vasculares
Trombosis venosa profunda*c d %
8 ( 4)
< 1%
5 ( 2)
< 1%
7 ( 2)
< 1%
4 ( 1)
< 1%
Neoplasias benignas, malignas y no especificadas (incluidos quistes y pólipos)
Síndrome mielodisplásico c d e
5 ( 2)
0 ( 0)
< 1%
0 ( 0)
3 ( 1)
0 ( 0)
< 1%
0 ( 0)
Después de al menos una terapia previa para MM:
Los datos se evaluaron de 703 pacientes en dos estudios que recibieron al menos una dosis de REVLIMID/dexametasona (353 pacientes) o placebo/dexametasona (350 pacientes).
En el grupo de tratamiento con REVLIMID/dexametasona, 269 pacientes (76%) tuvieron al menos una interrupción de la dosis con o sin una reducción de la dosis de REVLIMID en comparación con 199 pacientes (57%) en el grupo de tratamiento con placebo/dexametasona. De estos pacientes que tuvieron una interrupción de la dosis con o sin una reducción de la dosis, el 50% en el grupo de tratamiento con REVLIMID/dexametasona tuvo al menos una interrupción de la dosis adicional con o sin una reducción de la dosis en comparación con el 21% en el grupo de tratamiento con placebo/dexametasona. La mayoría de las reacciones adversas y las reacciones adversas de Grado 3/4 fueron más frecuentes en los pacientes que recibieron la combinación de REVLIMID/dexametasona en comparación con placebo/dexametasona.
Las Tablas 6, 7 y 8 resumen las reacciones adversas notificadas para los grupos REVLIMID/dexametasona y placebo/dexametasona.
Tabla 6: Reacciones adversas notificadas en ≥5% de los pacientes y con una diferencia ≥2% en la proporción de pacientes con MM entre los grupos REVLIMID/dexametasona y placebo/dexametasona
Sistema corporal
Reacción adversa
REVLIMID/Dex
(N=353)
n (%)
Placebo/Dex
(N=350)
n (%)
Trastornos de la sangre y del sistema linfático
Neutropenia%
149 (42)
22 ( 6)
Anemia@
111 (31)
83 (24)
Trombocitopenia@
76 (22)
37 (11)
Leucopenia
28 ( 8)
4 ( 1)
Linfopenia
19 ( 5)
5 ( 1)
Trastornos generales y condiciones del lugar de administración
Fatiga
155 (44)
146 (42)
Pirexia
97 (27)
82 (23)
Edema periférico
93 (26)
74 (21)
Dolor de pecho
29 ( 8)
20 ( 6)
Letargo
24 ( 7)
8 ( 2)
Trastornos gastrointestinales
Estreñimiento
143 (41)
74 (21)
Diarrea@
136 (39)
96 (27)
Náuseas@
92 (26)
75 (21)
Vómitos@
43 (12)
33 ( 9)
Dolor abdominal@
35 ( 10)
22 ( 6)
Boca seca
25 ( 7)
13 ( 4)
Trastornos musculoesqueléticos y del tejido conjuntivo
Calambre muscular
118 (33)
74 (21)
Dolor de espalda
91 (26)
65 (19)
Dolor óseo
48 (14)
39 (11)
Dolor en la extremidad
42 (12)
32 ( 9)
Trastornos del sistema nervioso
Mareo
82 (23)
59 (17)
Temblor
75 (21)
26 ( 7)
Disgeusia
54 (15)
34 ( 10)
Hipoestesia
36 (10)
25 ( 7)
Neuropatíaa
23 ( 7)
13 ( 4)
Trastornos respiratorios, torácicos y mediastínicos
Disnea
83 (24)
60 (17)
Nasofaringitis
62 (18)
31 ( 9)
Faringitis
48 (14)
33 ( 9)
Bronquitis
40 (11)
30 ( 9)
Infeccionesb e infestaciones
Infección del tracto respiratorio superior
87 (25)
55 (16)
Neumonía@
48 (14)
29 ( 8)
Infección del tracto urinario
30 ( 8)
19 ( 5)
Sinusitis
26 ( 7)
16 ( 5)
Trastornos de la piel y del tejido subcutáneo
Erupciónc
75 (21)
33 ( 9)
Aumento de la sudoración
35 ( 10)
25 ( 7)
Piel seca
33 ( 9)
14 ( 4)
Prurito
27 ( 8)
18 ( 5)
Trastornos del metabolismo y la nutrición
Anorexia
55 (16)
34 ( 10)
Hipopotasemia
48 (14)
21 ( 6)
Hipocalcemia
31 ( 9)
10 ( 3)
Disminución del apetito
24 ( 7)
14 ( 4)
Deshidratación
23 ( 7)
15 ( 4)
Hipomagnesemia
24 ( 7)
10 ( 3)
Investigaciones
Pérdida de peso
69 (20)
52 (15)
Trastornos oculares
Visión borrosa
61 (17)
40 (11)
Trastornos vasculares
Trombosis venosa profunda%
33 ( 9)
15 ( 4)
Hipertensión
28 ( 8)
20 ( 6)
Hipotensión
25 ( 7)
15 ( 4)
Tabla 7: Reacciones adversas de grado 3/4 notificadas en ≥2% de los pacientes y con una diferencia ≥1% en la proporción de pacientes con MM entre los grupos REVLIMID/dexametasona y placebo/dexametasona
Sistema corporal
Reacción adversa
REVLIMID/Dex
(N=353)
n (%)
Placebo/Dex
(N=350)
n (%)
Trastornos de la sangre y del sistema linfático
Neutropenia%
118 (33)
12 ( 3)
Trombocitopenia@
43 (12)
22 ( 6)
Anemia@
35 ( 10)
20 ( 6)
Leucopenia
14 ( 4)
< 1%
Linfopenia
10 ( 3)
4 ( 1)
Neutropenia febril%
8 ( 2)
0 ( 0)
Trastornos generales y condiciones del lugar de administración
Fatiga
23 ( 7)
17 ( 5)
Trastornos vasculares
Trombosis venosa profunda%
29 ( 8)
12 ( 3)
Infecciones e infestaciones
Neumonía@
30 ( 8)
19 ( 5)
Infección del tracto urinario
5 ( 1)
< 1%
Trastornos del metabolismo y la nutrición
Hipokalemia
17 ( 5)
5 ( 1)
Hipocalcemia
13 ( 4)
6 ( 2)
Hipofosfatemia
9 ( 3)
0 ( 0)
Trastornos respiratorios, torácicos y mediastínicos
Embolia pulmonar@
14 ( 4)
< 1%
Dificultad respiratoria@
4 ( 1)
0 ( 0)
Trastornos musculoesqueléticos y del tejido conjuntivo
Debilidad muscular
20 ( 6)
10 ( 3)
Trastornos gastrointestinales
Diarrea@
11 ( 3)
4 ( 1)
Estreñimiento
7 ( 2)
< 1%
Náuseas@
6 ( 2)
< 1%
Trastornos cardíacos
Fibrilación auricular@
13 ( 4)
4 ( 1)
Taquicardia
6 ( 2)
< 1%
Insuficiencia cardíaca congestiva@
5 ( 1)
< 1%
Trastornos del sistema nervioso
Síncope
10 ( 3)
< 1%
Mareos
7 ( 2)
< 1%
Trastornos oculares
Catarata
6 ( 2)
< 1%
Catarata unilateral
5 ( 1)
0 ( 0)
Trastorno psiquiátrico
Depresión
10 ( 3)
6 ( 2)
Tabla 8: Reacciones adversas graves notificadas en ≥1% de los pacientes y con una diferencia ≥1% en la proporción de pacientes con MM entre los grupos REVLIMID/dexametasona y placebo/dexametasona
Sistema corporal
Reacción adversa
REVLIMID/Dex
(N=353)
n (%)
Placebo/Dex
(N=350)
n (%)
Para las Tablas 6, 7 y 8 anteriores: @ – reacciones adversas en las que al menos una tuvo como resultado un desenlace fatal. % – reacciones adversas en las que al menos una se consideró que ponía en peligro la vida (si el resultado de la reacción fue la muerte, se incluye con los casos de muerte).
Trastornos de la sangre y del sistema linfático
Neutropenia febril%
6 ( 2)
0 ( 0)
Trastornos vasculares
Trombosis venosa profunda%
26 ( 7)
11 ( 3)
Infecciones e infestaciones
Neumonía@
33 ( 9)
21 ( 6)
Trastornos respiratorios, torácicos y mediastínicos
Embolia pulmonar@
13 ( 4)
< 1%
Trastornos cardíacos
Fibrilación auricular@
11 ( 3)
< 1%
Insuficiencia cardíaca congestiva@
5 ( 1)
0 ( 0)
Trastornos del sistema nervioso
Accidente cerebrovascular@
7 ( 2)
< 1%
Trastornos gastrointestinales
Diarrea @
6 ( 2)
< 1%
Trastornos musculoesqueléticos y del tejido conjuntivo
Dolor óseo
4 ( 1)
0 ( 0)
La duración media de la exposición entre los pacientes tratados con REVLIMID/dexametasona fue de 44 semanas, mientras que la duración media de la exposición entre los pacientes tratados con placebo/dexametasona fue de 23 semanas. Esto debe tenerse en cuenta al comparar la frecuencia de reacciones adversas entre los dos grupos de tratamiento REVLIMID/dexametasona frente a placebo/dexametasona.
El TVE y el TAE aumentan en los pacientes tratados con REVLIMID.
La trombosis venosa profunda (TVP) se notificó como una reacción adversa al fármaco grave (7,4%) o severa (8,2%) a una tasa más alta en el grupo REVLIMID/dexametasona en comparación con el 3,1% y el 3,4% en el grupo placebo/dexametasona, respectivamente, en los 2 estudios en pacientes con al menos 1 terapia previa con interrupciones debido a reacciones adversas de TVP notificadas a tasas comparables entre los grupos. En el estudio NDMM, la TVP se notificó como una reacción adversa (todos los grados: 10,3%, 7,2%, 4,1%), como una reacción adversa grave (3,6%, 2,0%, 1,7%) y como una reacción adversa de grado 3/4 (5,6%, 3,7%, 2,8%) en los brazos Rd Continuo, Rd18 y MPT, respectivamente. Las interrupciones y las reducciones de dosis debido a reacciones adversas de TVP se notificaron a tasas comparables entre los brazos Rd Continuo y Rd18 (ambos <1%). La interrupción del tratamiento con REVLIMID debido a reacciones adversas de TVP se notificó a tasas comparables entre los brazos Rd Continuo (2,3%) y Rd18 (1,5%). La embolia pulmonar (EP) se notificó como una reacción adversa al fármaco grave (3,7%) o de grado 3/4 (4,0%) a una tasa más alta en el grupo REVLIMID/dexametasona en comparación con el 0,9% (grave o de grado 3/4) en el grupo placebo/dexametasona en los 2 estudios en pacientes con, al menos 1 terapia previa, con interrupciones debido a reacciones adversas de EP notificadas a tasas comparables entre los grupos. En el estudio NDMM, la frecuencia de reacciones adversas de EP fue similar entre los brazos Rd Continuo, Rd18 y MPT para reacciones adversas (todos los grados: 3,9%, 3,3% y 4,3%, respectivamente), reacciones adversas graves (3,8%, 2,8% y 3,7%, respectivamente) y reacciones adversas de grado 3/4 (3,8%, 3,0% y 3,7%, respectivamente).
El infarto de miocardio se notificó como una reacción adversa al fármaco grave (1,7%) o severa (1,7%) a una tasa más alta en el grupo REVLIMID/dexametasona en comparación con el 0,6% y el 0,6% respectivamente en el grupo placebo/dexametasona. La interrupción debido a reacciones adversas de IM (incluido el agudo) fue del 0,8% en el grupo REVLIMID/dexametasona y ninguna en el grupo placebo/dexametasona. En el estudio NDMM, el infarto de miocardio (incluido el agudo) se notificó como una reacción adversa (todos los grados: 2,4%, 0,6% y 1,1%), como una reacción adversa grave, (2,3%, 0,6% y 1,1%), o como una reacción adversa severa (1,9%, 0,6% y 0,9%) en los brazos Rd Continuo, Rd18 y MPT, respectivamente.
El accidente cerebrovascular (ACV) se notificó como una reacción adversa al fármaco grave (2,3%) o severa (2,0%) en el grupo REVLIMID/dexametasona en comparación con el 0,9% y el 0,9% respectivamente en el grupo placebo/dexametasona. La interrupción debido a accidente cerebrovascular (ACV) fue del 1,4% en el grupo REVLIMID/dexametasona y del 0,3% en el grupo placebo/dexametasona. En el estudio NDMM, el ACV se notificó como una reacción adversa (todos los grados: 0,8%, 0,6% y 0,6%), como una reacción adversa grave (0,8%, 0,6% y 0,6%), o como una reacción adversa severa (0,6%, 0,6%, 0,2%) en los brazos Rd Continuo, Rd18 y MPT respectivamente.
Otras reacciones adversas: después de al menos una terapia previa para el MM
En estos 2 estudios, se notificaron las siguientes reacciones adversas al fármaco (RAD) no descritas anteriormente que ocurrieron a una tasa ≥1% y al menos el doble de la tasa porcentual del placebo:
Trastornos de la sangre y del sistema linfático: pancitopenia, anemia hemolítica autoinmune
Trastornos cardíacos: bradicardia, infarto de miocardio, angina de pecho
Trastornos generales y condiciones del lugar de administración: malestar
Investigaciones: pruebas de función hepática anormales, aumento de la alanina aminotransferasa
Trastornos del sistema nervioso: isquemia cerebral
Trastornos psiquiátricos: cambios de humor, alucinaciones, pérdida de la libido
Trastornos del aparato reproductor y de la mama: disfunción eréctil
Trastornos respiratorios, torácicos y mediastínicos: tos, ronquera
Trastornos de la piel y del tejido subcutáneo: exantema, hiperpigmentación de la piel
Síndromes mielodisplásicos:
Un total de 148 pacientes recibieron al menos 1 dosis de 10 mg de REVLIMID en el estudio clínico de MDS del 5q. Se notificó al menos una reacción adversa en todos los 148 pacientes que fueron tratados con la dosis inicial de 10 mg de REVLIMID. Las reacciones adversas más frecuentes fueron las relacionadas con trastornos de la sangre y del sistema linfático, trastornos de la piel y del tejido subcutáneo, trastornos gastrointestinales y trastornos generales y condiciones del lugar de administración.
La trombocitopenia (61,5%; 91/148) y la neutropenia (58,8%; 87/148) fueron las reacciones adversas más frecuentes. Las siguientes reacciones adversas más comunes observadas fueron diarrea (48,6%; 72/148), prurito (41,9%; 62/148), erupción cutánea (35,8%; 53/148) y fatiga (31,1%; 46/148). La Tabla 9 resume las reacciones adversas que se notificaron en ≥ 5% de los pacientes tratados con REVLIMID en el estudio clínico de MDS del 5q. La Tabla 10 resume las reacciones adversas de grado 3 y grado 4 más frecuentes observadas independientemente de la relación con el tratamiento con REVLIMID. En los estudios de un solo brazo realizados, a menudo no es posible distinguir las reacciones adversas que están relacionadas con el fármaco y las que reflejan la enfermedad subyacente del paciente.
Tabla 9: Resumen de las reacciones adversas notificadas en ≥5% de los pacientes tratados con REVLIMID en el estudio clínico de MDS del 5q
a El sistema corporal y las reacciones adversas se codifican utilizando el diccionario MedDRA. El sistema corporal y las reacciones adversas se enumeran en orden descendente de frecuencia para la columna General. Un paciente con múltiples ocurrencias de una reacción adversa se cuenta solo una vez bajo el Sistema corporal/Reacción adversa aplicable.
Sistema corporal
Reacción adversa a
10 mg General
(N=148)
Pacientes con al menos una reacción adversa
148 (100)
Trastornos de la sangre y del sistema linfático
Trombocitopenia
91 (61)
Neutropenia
87 (59)
Anemia
17 (11)
Leucopenia
12 (8)
Neutropenia febril
8 (5)
Trastornos de la piel y del tejido subcutáneo
Prurito
62 (42)
Erupción cutánea
53 (36)
Piel seca
21 (14)
Contusión
12 (8)
Sudoración nocturna
12 (8)
Aumento de la sudoración
10 (7)
Equimosis
8 (5)
Eritema
8 (5)
Trastornos gastrointestinales
Diarrea
72 (49)
Estreñimiento
35 (24)
Náuseas
35 (24)
Dolor abdominal
18 (12)
Vómitos
15 (10)
Dolor abdominal superior
12 (8)
Boca seca
10 (7)
Heces blandas
9 (6)
Trastornos respiratorios, torácicos y mediastínicos
Nasofaringitis
34 (23)
Tos
29 (20)
Disnea
25 (17)
Faringitis
23 (16)
Epistaxis
22 (15)
Disnea de esfuerzo
10 (7)
Rinitis
10 (7)
Bronquitis
9 (6)
Trastornos generales y condiciones del lugar de administración
Fatiga
46 (31)
Pirexia
31 (21)
Edema periférico
30 (20)
Astenia
22 (15)
Edema
15 (10)
Dolor
10 (7)
Escalofríos
9 (6)
Dolor de pecho
8 (5)
Trastornos músculo esqueléticos y del tejido conjuntivo
Artralgia
32 (22)
Dolor de espalda
31 (21)
Calambres musculares
27 (18)
Dolor en la extremidad
16 (11)
Mialgia
13 (9)
Hinchazón periférica
12 (8)
Trastornos del sistema nervioso
Mareos
29 (20)
Dolor de cabeza
29 (20)
Hipoestesia
10 (7)
Disgeusia
9 (6)
Neuropatía periférica
8 (5)
Infecciones e infestaciones
Infección de las vías respiratorias superiores
22 (15)
Neumonía
17 (11)
Infección del tracto urinario
16 (11)
Sinusitis
12 (8)
Celulitis
8 (5)
Trastornos del metabolismo y la nutrición
Hipokalemia
16 (11)
Anorexia
15 (10)
Hipomagnesemia
9 (6)
Investigaciones
Aumento de la alanina aminotransferasa
12 (8)
Trastornos psiquiátricos
Insomnio
15 (10)
Depresión
8 (5)
Trastornos renales y urinarios
Disuria
10 (7)
Trastornos vasculares
Hipertensión
9 (6)
Trastornos endocrinos
Hipotiroidismo adquirido
10 (7)
Trastornos cardíacos
Palpitaciones
8 (5)
Tabla 10: Reacciones adversas de Grado 3 y 4 observadas con mayor frecuencia 1 Independientemente de la relación con el tratamiento con el fármaco del estudio en el estudio clínico de MDS del 5q del
1 Reacciones adversas con una frecuencia ≥1% en el grupo general de 10 mg. Los grados 3 y 4 se basan en la versión 2 de los Criterios Comunes de Toxicidad del Instituto Nacional del Cáncer. 2 Las reacciones adversas se codifican utilizando el diccionario MedDRA. Un paciente con múltiples ocurrencias de una reacción adversa se cuenta solo una vez en la categoría de reacción adversa.
Reacciones adversas 2
10 mg
(N=148)
Pacientes con al menos un AE de Grado 3/4
131 (89)
Neutropenia
79 (53)
Trombocitopenia
74 (50)
Neumonía
11 (7)
Erupción
10 (7)
Anemia
9 (6)
Leucopenia
8 (5)
Fatiga
7 (5)
Disnea
7 (5)
Dolor de espalda
7 (5)
Neutropenia febril
6 (4)
Náuseas
6 (4)
Diarrea
5 (3)
Pirexia
5 (3)
Sepsis
4 (3)
Mareo
4 (3)
Granulocitopenia
3 (2)
Dolor de pecho
3 (2)
Embolia pulmonar
3 (2)
Dificultad respiratoria
3 (2)
Prurito
3 (2)
Pancytopenia
3 (2)
Calambre muscular
3 (2)
Infección del tracto respiratorio
2 (1)
Infección del tracto respiratorio superior
2 (1)
Astenia
2 (1)
Insuficiencia multiorgánica
2 (1)
Epistaxis
2 (1)
Hipoxia
2 (1)
Derrame pleural
2 (1)
Neumonitis
2 (1)
Hipertensión pulmonar
2 (1)
Vómitos
2 (1)
Sudoración aumentada
2 (1)
Artralgia
2 (1)
Dolor en la extremidad
2 (1)
Dolor de cabeza
2 (1)
Síncope
2 (1)
En otros estudios clínicos de REVLIMID en pacientes con SMD, se informaron las siguientes reacciones adversas graves (independientemente de la relación con el tratamiento con el fármaco del estudio) que no se describen en la Tabla 9 o 10:
Trastornos de la sangre y del sistema linfático: anemia hemolítica de tipo cálido, infarto esplénico, depresión de la médula ósea, coagulopatía, hemólisis, anemia hemolítica, anemia refractaria
Trastornos gastrointestinales: hemorragia gastrointestinal, colitis isquémica, perforación intestinal, hemorragia rectal, pólipo colónico, diverticulitis, disfagia, gastritis, gastroenteritis, enfermedad por reflujo gastroesofágico, hernia inguinal obstructiva, síndrome del intestino irritable, melena, pancreatitis por obstrucción biliar, pancreatitis, absceso perirrectal, obstrucción del intestino delgado, hemorragia gastrointestinal alta
Trastornos generales y condiciones del lugar de administración: progresión de la enfermedad, caída, marcha anormal, pirexia intermitente, nódulo, escalofríos, muerte súbita
Trastornos del sistema inmunitario: hipersensibilidad
Infecciones e infestaciones: infección bacteriemia, infección de línea central, infección por clostridios, infección del oído, Enterobacter sepsis, infección micótica, infección viral por herpes NOS, influenza, infección renal, Klebsiella sepsis, neumonía lobar, infección localizada, infección oral, Pseudomonas infección, shock séptico, sinusitis aguda, sinusitis, Staphylococcal infección, urosepsis
Lesiones, envenenamiento y complicaciones de procedimientos: fractura de fémur, reacción transfusional, fractura vertebral cervical, fractura del cuello femoral, fractura de pelvis, fractura de cadera, sobredosis, hemorragia postprocedimiento, fractura de costilla, accidente de tráfico, fractura por compresión espinal
Investigaciones: aumento de la creatinina en sangre, disminución de la hemoglobina, pruebas de función hepática anormales, aumento de la troponina I
Trastornos del metabolismo y la nutrición: deshidratación, gota, hipernatremia, hipoglucemia
Trastornos musculoesqueléticos y del tejido conjuntivo: artritis, artritis agravada, artritis gotosa, dolor de cuello, condrocalcinosis pirofosfato
Neoplasias benignas, malignas y no especificadas: leucemia aguda, leucemia mieloide aguda, carcinoma broncoalveolar, cáncer de pulmón metastásico, linfoma, cáncer de próstata metastásico
Trastornos del sistema nervioso: accidente cerebrovascular, afasia, infarto cerebeloso, infarto cerebral, disminución del nivel de conciencia, disartria, migraña, compresión de la médula espinal, hemorragia subaracnoidea, ataque isquémico transitorio
Trastornos psiquiátricos: estado confusional
Trastornos renales y urinarios: insuficiencia renal, hematuria, insuficiencia renal aguda, azotemia, cálculo ureteral, masa renal
Trastornos del aparato reproductor femenino y de la mama: dolor pélvico
Trastornos respiratorios, torácicos y mediastínicos: bronquitis, exacerbación de la enfermedad pulmonar obstructiva crónica, insuficiencia respiratoria, disnea exacerbada, enfermedad pulmonar intersticial, infiltración pulmonar, sibilancias
Trastornos de la piel y del tejido subcutáneo: dermatosis neutófila febril aguda
Trastornos del sistema vascular: trombosis venosa profunda, hipotensión, trastorno aórtico, isquemia, tromboflebitis superficial, trombosis
Linfoma de células del manto:
En el ensayo de MCL, un total de 134 pacientes recibieron al menos 1 dosis de REVLIMID. Su edad media fue de 67 (rango 43-83) años, 128/134 (96%) eran caucásicos, 108/134 (81%) eran hombres y 82/134 (61%) tenían una duración de MCL de al menos 3 años.
La Tabla 11 resume las reacciones adversas más frecuentes observadas independientemente de la relación con el tratamiento con REVLIMID. En los 134 pacientes tratados en este estudio, la duración media del tratamiento fue de 95 días (1-1002 días). Setenta y ocho pacientes (58%) recibieron 3 o más ciclos de terapia, 53 pacientes (40%) recibieron 6 o más ciclos, y 26 pacientes (19%) recibieron 12 o más ciclos. Setenta y seis pacientes (57%) experimentaron al menos una interrupción de la dosis debido a reacciones adversas, y 51 pacientes (38%) experimentaron al menos una reducción de la dosis debido a reacciones adversas. Veintiséis pacientes (19%) interrumpieron el tratamiento debido a reacciones adversas.
Tabla 11: Incidencia de reacciones adversas (≥10%) o AE de grado 3/4 (en al menos 2 pacientes) en el linfoma de células del manto
1-AE del ensayo de MCL – Todas las AE emergentes del tratamiento con ≥10% de los sujetos. 2-AE de grado 3/4 del ensayo de MCL – Todas las AE de grado 3/4 emergentes del tratamiento en 2 o más sujetos. $-AE graves del ensayo de MCL – Todas las SAE emergentes del tratamiento en 2 o más sujetos. @ – Reacciones adversas donde al menos una resultó en un desenlace fatal. % – Reacciones adversas donde al menos una se consideró que ponía en peligro la vida (si el desenlace del evento fue la muerte, se incluye con los casos de muerte). # – Todas las reacciones adversas bajo el Sistema Corporal de Infecciones, excepto las infecciones raras de interés para la salud pública, se considerarán incluidas. + – Todas las reacciones adversas bajo el HLT de Erupción se considerarán incluidas.
Sistema Corporal
Reacción Adversa
Todas las reacciones adversas1 (N=134) n (%)
Reacciones adversas de grado 3/42 (N=134) n (%)
Trastornos generales y condiciones del lugar de administración
Fatiga
45 (34)
9 (7)
Pirexia$
31 (23)
3 (2)
Edema periférico
21 (16)
0
Astenia$
19 (14)
4 (3)
Deterioro general de la salud física
3 (2)
2 (1)
Trastornos gastrointestinales
Diarrea$
42 (31)
8 (6)
Náuseas$
40 (30)
1 (<1)
Estreñimiento
21 (16)
1 (<1)
Vómitos$
16 (12)
1 (<1)
Dolor abdominal$
13 (10)
5 ( 4)
Trastornos musculoesqueléticos y del tejido conjuntivo
Dolor de espalda
18 (13)
2 (1)
Espasmos musculares
17 (13)
1 (<1)
Artralgia
11 (8)
2 (1)
Debilidad muscular$
8 (6)
2 ( 1)
Trastornos respiratorios, torácicos y mediastínicos
Tos
38 (28)
1 (<1)
Disnea$
24 (18)
8 (6)
Derrame pleural
10 (7)
2 (1)
Hipoxia
3 (2)
2 (1)
Embolia pulmonar
3 (2)
2 ( 1)
Dificultad respiratoria$
2 (1)
2 (1)
Dolor orofaríngeo
13 (10)
0
Infecciones e infestaciones
Neumonía@ $
19 (14)
12 (9)
Infección de las vías respiratorias altas
17 (13)
0
Celulitis$
3 (2)
2 (1)
Bacteriemia$
2 (1)
2 (1)
Sepsis estafilocócica$
2 (1)
2 (1)
Infección del tracto urinario$
5 (4)
2 (1)
Trastornos de la piel y del tejido subcutáneo
Erupción cutánea +
30 (22)
2 (1)
Prurito
23 (17)
1 (<1)
Trastornos de la sangre y del sistema linfático
Neutropenia
65 (49)
58 (43)
Trombocitopenia% $
48 (36)
37 (28)
Anemia$
41 (31)
15 (11)
Leucopenia$
20 (15)
9 (7)
Linfopenia
10 ( 7)
5 (4)
Neutropenia febril$
8 (6)
8 (6)
Trastornos del metabolismo y la nutrición
Disminución del apetito
19 (14)
1 (<1)
Hipokalemia
17 (13)
3 (2)
Deshidratación$
10 (7)
4 (3)
Hipocalcemia
4 (3)
2 (1)
Hiponatremia
3 (2)
3 (2)
Trastornos renales y urinarios
Insuficiencia renal$
5 (4)
2 (1)
Trastornos vasculares
Hipotensión@ $
9 (7)
4 (3)
Trombosis venosa profunda$
5 (4)
5 (4)
Neoplasias benignas, malignas y no especificadas (incluidos quistes y pólipos)
Brote tumoral
13 (10)
0
Carcinoma de células escamosas de la piel$
4 (3)
4 (3)
Investigaciones
Pérdida de peso
17 (13)
0
Las siguientes reacciones adversas que se han producido en otras indicaciones, incluido otro estudio de MCL y no descritas anteriormente, se han notificado (1%-10%) en pacientes tratados con REVLIMID en monoterapia para el linfoma de células del manto.
Trastorno cardíaco: Insuficiencia cardíaca
Trastornos del oído y el laberinto: Vértigo
Trastornos generales y condiciones del lugar de administración: Escalofríos
Infecciones e infestaciones: Infección del tracto respiratorio, sinusitis, nasofaringitis, herpes oral
Trastornos musculoesqueléticos y del tejido conjuntivo: Dolor en las extremidades
Trastornos del sistema nervioso: Disgeusia, dolor de cabeza, neuropatía periférica, letargo
Trastornos psiquiátricos: Insomnio
Trastornos de la piel y del tejido subcutáneo: Piel seca, sudoración nocturna
Las siguientes reacciones adversas graves no descritas anteriormente y notificadas en 2 o más pacientes tratados con REVLIMID en monoterapia para el linfoma de células del manto.
Trastornos de la sangre y del sistema linfático: Neutropenia
Trastorno cardíaco: Infarto de miocardio (incluido el IM agudo), taquicardia supraventricular
Infecciones e infestaciones:Clostridium difficile colitis, sepsis
Neoplasias benignas, malignas e indeterminadas (incluidos quistes y pólipos): Carcinoma de células basales
La seguridad de REVLIMID/rituximab se evaluó en 398 pacientes con linfoma folicular o linfoma de la zona marginal previamente tratados en dos ensayos clínicos; AUGMENT (N=176) y MAGNIFY (N=222) [ver Estudios clínicos (14.4)]. Los sujetos tenían 18 años o más, tenían un ECOG PS ≤2, ANC ≥1.000 células/mm3 y plaquetas ≥ 75.000/mm3 (a menos que fuera secundario a la afectación de la médula ósea por el linfoma), hemoglobina ≥8 g/dL, AST y ALT ≤ 3 x ULN (a menos que se documentara la afectación hepática por el linfoma, y aclaramiento de creatinina de ≥ 30 ml/min. Los sujetos con VIH activo, hepatitis B o C no eran elegibles.
En el ensayo AUGMENT, los pacientes recibieron REVLIMID 20 mg diarios por vía oral los días 1 a 21 de cada ciclo de 28 días con rituximab 375 mg/m2 semanal (días 1, 8, 15 y 22 en el ciclo 1) y luego el día 1 de los ciclos 2-5 (n=176) o placebo con rituximab 375 mg/m2 semanal (días 1, 8, 15 y 22 en el ciclo 1) y luego el día 1 de los ciclos 2-5 (n=180) durante un máximo de 12 ciclos. En el ensayo MAGNIFY, los pacientes recibieron REVLIMID 20 mg por vía oral diariamente, días 1-21 de cada ciclo de 28 días con rituximab 375 mg/m2 semanal (días 1, 8, 15 y 22 en el ciclo 1) y luego el día 1 de los ciclos 3, 5, 7, 9 y 11 en la fase de inducción del ensayo (n=222). En el ensayo AUGMENT, el 88,1% de los pacientes completaron al menos 6 ciclos de REVLIMID/rituximab, y el 71% de los pacientes completaron 12 ciclos. En el ensayo MAGNIFY en curso, al 1 de mayo de 2017, el 62,2% de los pacientes completaron al menos 6 ciclos de REVLIMID/rituximab, y el 30,6% de los pacientes completaron 12 ciclos.
En ambos ensayos clínicos (AUGMENT y MAGNIFY), los pacientes tenían una edad media de 64,5 años (26 a 91); el 49% eran hombres; y el 81% eran blancos.
Se produjeron reacciones adversas fatales en 6 pacientes (1,5%) que recibieron REVLIMID/rituximab. Las reacciones adversas fatales (1 cada una) incluyeron paro cardiorrespiratorio, arritmia, insuficiencia cardiopulmonar, síndrome de disfunción multiorgánica, sepsis e insuficiencia renal aguda. Las reacciones adversas graves se produjeron en el 26% de los pacientes que recibieron REVLIMID/rituximab en AUGMENT y en el 29% en MAGNIFY. La reacción adversa grave más frecuente que se produjo en ≥ 2,5% de los pacientes en el brazo de REVLIMID/rituximab fue la neutropenia febril (3%). La interrupción permanente de REVLIMID o rituximab debido a una reacción adversa se produjo en el 14,6% de los pacientes en el brazo de REVLIMID/rituximab. La reacción adversa más común (en al menos el 1%) que requirió la interrupción permanente de REVLIMID o rituximab fue la neutropenia (4,8%).
Las reacciones adversas más comunes que se produjeron en al menos el 20% de los sujetos fueron: neutropenia (48%), fatiga (37%), diarrea (32%), estreñimiento (27%), náuseas (21%) y tos (20%).
Tabla 12: Todas las reacciones adversas de cualquier grado (≥5%) o reacciones adversas de grado 3/4 (≥1%) en pacientes con FL y MZL con una diferencia entre brazos de >1% en comparación con el brazo de control en el ensayo AUGMENT
Nota: Las reacciones adversas se codifican según el sistema corporal/reacción adversa utilizando MedDRA 21. Un paciente con múltiples ocurrencias de una reacción adversa se cuenta solo una vez bajo el Sistema corporal/Reacción adversa aplicable. 1 Todos los AE emergentes del tratamiento en al menos el 5% de los pacientes en el grupo REVLIMID + rituximab y al menos 1% de frecuencia (%) mayor que el grupo rituximab + placebo (brazo de control). 2 Todos los AE emergentes del tratamiento de grado 3 o 4 en al menos el 1% de los pacientes en el grupo REVLIMID + rituximab y al menos 1% de frecuencia (%) mayor que el grupo rituximab + placebo (brazo de control). 3 Todos los AE emergentes del tratamiento graves en al menos el 1% de los pacientes en el grupo REVLIMID + rituximab y al menos 1% de frecuencia (%) mayor que el grupo rituximab + placebo (brazo de control). $ ADR grave notificada. @ – reacciones adversas en las que al menos una resultó en un desenlace fatal. % – reacciones adversas en las que al menos una se consideró que ponía en peligro la vida (si el desenlace de la reacción fue la muerte, se incluye con los casos de muerte). *Reacciones adversas para términos combinados de ADR (basados en PT relevantes de TEAE [según MedDRA versión 21.0]): a “Eventos tromboembólicos” el término combinado incluye los siguientes PT: embolia pulmonar, trombosis venosa profunda, accidente cerebrovascular, embolia y trombosis. b “Tos” el término combinado de AE incluye los siguientes PT: tos y tos productiva.
c “Dolor abdominal” el término combinado de AE incluye los siguientes PT: dolor abdominal y dolor abdominal superior. d “Erupción” el término combinado de AE incluye los siguientes PT: erupción maculo-papular, erupción eritematosa, erupción macular, erupción papular, erupción pruriginosa y erupción generalizada. e “Prurito” el término combinado de AE incluye los siguientes PT: prurito, prurito generalizado, erupción pruriginosa y prurito alérgico.
Todas las reacciones adversas 1
Reacciones adversas de grado 3 / 4 2
Sistema corporal
Reacción adversa*
Brazo de REVLIMID + Rituximab
(N=176)
n (%)
Rituximab + Placebo (Brazo de control)
(N=180)
n (%)
Brazo de REVLIMID + Rituximab
(N=176)
n (%)
Rituximab + Placebo (Brazo de control)
(N=180)
n (%)
Infecciones e infestaciones
Infección del tracto respiratorio superior
32 (18)
23 (13)
2 (1.1)
4 (2.2)
Influenza %
17 (10)
8 (4.4)
1 (< 1)
0 (0)
Neumonía 3,$,%
13 (7)
6 (3.3)
6 (3.4)
4 (2.2)
Sinusitis
13 (7)
5 (2.8)
0 (0)
0 (0)
Infección del tracto urinario$
13 (7)
7 (3.9)
1 (< 1)
1 (< 1)
Bronquitis
8 (4.5)
6 (3.3)
2 (1.1)
0 (0)
Gastroenteritis $
6 (3.4)
4 (2.2)
2 (1.1)
0 (0)
Neoplasias benignas, malignas e indeterminadas (incluidos quistes y pólipos)
Brote tumoral $
19 (11)
1 (< 1)
1 (< 1)
0 (0)
Trastornos de la sangre y del sistema linfático
Neutropenia 3,$, %
102 (58)
40 (22)
88 (50)
23 (13)
Leucopenia $,%
36 (20)
17 (9)
12 (7)
3 (1.7)
Anemia 3,$
28 (16)
8 (4.4)
8 (4.5)
1 (< 1)
Trombocitopenia 3,$,%
26 (15)
8 (4.4)
4 (2.3)
2 (1.1)
Linfopenia
8 (4.5)
14 (8)
5 (2.8)
2 (1.1)
Neutropenia febril 3,$,%
5 (2.8)
1 (< 1)
5 (2.8)
1 (< 1)
Trastornos del metabolismo y la nutrición
Disminución del apetito
23 (13)
11 (6)
2 (1.1)
0 (0)
Hipokalemia %
14 (8)
5 (2.8)
4 (2.3)
0 (0)
Hiperuricemia
10 (6)
8 (4.4)
1 (< 1)
1 (< 1)
Trastornos del sistema nervioso
Dolor de cabeza
26 (15)
17 (9)
1 (< 1)
0 (0)
Mareos
15 (9)
9 (5)
0 (0)
0 (0)
Trastornos vasculares
Hipotensión %
9 (5)
1 (< 1)
1 (< 1)
0 (0)
Eventos tromboembólicos a,$
8 (4.5)
2 (1.1)
4 (2.3)
2 (1.1)
Trastornos respiratorios, torácicos y mediastínicos
Tos b
43 (24)
35 (19)
1 (< 1)
0 (0)
Disnea $
19 (11)
8 (4.4)
2 (1.1)
1 (< 1)
Dolor orofaríngeo
10 (6)
8 (4.4)
0 (0)
0 (0)
Embolia pulmonar 3,$
4 (2.3)
1 (< 1)
4 (2.3)
1 (< 1)
Enfermedad pulmonar obstructiva crónica $
3 (1.7)
0 (0)
2 (1.1)
0 (0)
Insuficiencia respiratoria 3,$
2 (1.1)
1 (< 1)
2 (1.1)
0 (0)
Trastornos gastrointestinales
Diarrea $,%
55 (31)
41 (23)
5 (2.8)
0 (0)
Estreñimiento
46 (26)
25 (14)
0 (0)
0 (0)
Dolor abdominal c ,$
32 (18)
20 (11)
2 (1.1)
0 (0)
Vómitos $
17 (10)
13 (7)
0 (0)
0 (0)
Dispepsia
16 (9)
5 (2.8)
0 (0)
0 (0)
Estomatitis
9 (5)
7 (3.9)
0 (0)
0 (0)
Trastornos de la piel y del tejido subcutáneo
Erupción cutánea $,d
39 (22)
14 (8)
5 (2.8)
2 (1.1)
Prurito $,e
36 (20)
9 (5)
2 (1.1)
0 (0)
Piel seca
9 (5)
6 (3.3)
0 (0)
0 (0)
Dermatitis acneiforme
8 (4.5)
0 (0)
2 (1.1)
0 (0)
Trastornos musculoesqueléticos y del tejido conjuntivo
Espasmos musculares
23 (13)
9 (5)
1 (< 1)
1 (< 1)
Dolor en las extremidades $
8 (4.5)
9 (5)
2 (1)
0 (0)
Trastornos renales
Lesión renal aguda 3,$,@,%
3 (1.7)
0 (0)
2 (1.1)
0 (0)
Trastornos cardíacos
Taquicardia supraventricular 3,$
2 (1.1)
0 (0)
2 (1.1)
0 (0)
Trastornos generales y condiciones del sitio de administración
Fatiga
38 (22)
33 (18)
2 (1.1)
1 (< 1)
Fiebre 3,$
37 (21)
27 (15)
1 (< 1)
3 (1.7)
Astenia $,%
24 (14)
19 (11)
2 (1.1)
1 (< 1)
Edema periférico $
23 (13)
16 (9)
0 (0)
0 (0)
Escalofríos
14 (8)
8 (4.4)
0 (0)
0 (0)
Malestar
13 (7)
10 (6)
0 (0)
0 (0)
Enfermedad similar a la gripe
9 (5)
7 (3.9)
0 (0)
0 (0)
Trastornos psiquiátricos
Insomnio
14 (8)
11 (6)
0 (0)
0 (0)
Pruebas de laboratorio
Alanina aminotransferasa aumentada
18 (10)
15 (8)
3 (1.7)
1 (< 1)
Recuento de glóbulos blancos disminuido
16 (9)
13 (7)
5 (2.8)
2 (1.1)
Recuento de linfocitos disminuido
12 (7)
12 (7)
6 (3.4)
2 (1.1)
Bilirrubina en sangre aumentada
10 (6)
0 (0)
0 (0)
0 (0)
Peso disminuido
12 (7)
2 (1.1)
0 (0)
0 (0)
6.2 Experiencia postcomercialización
Las siguientes reacciones adversas a los medicamentos se han identificado a partir de la experiencia postcomercialización mundial con REVLIMID. Debido a que estas reacciones se notifican voluntariamente de una población de tamaño incierto, no siempre es posible estimar de forma fiable su frecuencia o establecer una relación causal con la exposición al fármaco [ver Advertencias y precauciones Sección (5.8 a 5.11, y 5.13)]
Trastornos del sistema inmunitario: Angioedema, anafilaxia, enfermedad de injerto contra huésped aguda (después del trasplante hematopoyético alogénico), rechazo del trasplante de órganos sólidos
Infecciones e infestaciones: Reactivación viral (como el virus de la hepatitis B y el herpes zóster), leucoencefalopatía multifocal progresiva (PML)
Neoplasias benignas, malignas e indeterminadas (incluidos quistes y pólipos): Síndrome de lisis tumoral, reacción de brote tumoral
Trastornos respiratorios, torácicos y mediastínicos: Neumonitis
Trastornos de la piel y del tejido subcutáneo: Síndrome de Stevens-Johnson, necrólisis epidérmica tóxica, reacción medicamentosa con eosinofilia y síntomas sistémicos (DRESS)
7 INTERACCIONES MEDICAMENTOSAS
7.1 Digoxina
Cuando la digoxina se administró conjuntamente con dosis múltiples de REVLIMID (10 mg/día), la Cmax y el AUCinf de digoxina aumentaron un 14%. Controle periódicamente los niveles plasmáticos de digoxina, de acuerdo con el juicio clínico y en base a la práctica clínica estándar en pacientes que reciben este medicamento, durante la administración de REVLIMID.
7.2 Terapias concomitantes que pueden aumentar el riesgo de trombosis
Los agentes eritropoyéticos, u otros agentes que pueden aumentar el riesgo de trombosis, como las terapias que contienen estrógenos, deben utilizarse con precaución después de realizar una evaluación beneficio-riesgo en pacientes que reciben REVLIMID [ver Advertencias y precauciones (5.4)].
7.3 Warfarina
La administración conjunta de dosis múltiples de REVLIMID (10 mg/día) con una dosis única de warfarina (25 mg) no tuvo ningún efecto sobre la farmacocinética de la lenalidomida o la R- y S-warfarina. Se observaron los cambios esperados en las evaluaciones de laboratorio de PT e INR después de la administración de warfarina, pero estos cambios no se vieron afectados por la administración concomitante de REVLIMID. Se desconoce si existe una interacción entre la dexametasona y la warfarina. Se recomienda una estrecha vigilancia de PT e INR en pacientes con MM que toman warfarina de forma concomitante.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Registro de exposición durante el embarazo
Existe un registro de exposición durante el embarazo que monitoriza los resultados del embarazo en mujeres expuestas a REVLIMID durante el embarazo, así como en las parejas femeninas de pacientes masculinos que están expuestas a REVLIMID. Este registro también se utiliza para comprender la causa principal del embarazo. Informe cualquier sospecha de exposición fetal a REVLIMID a la FDA a través del programa MedWatch al 1-800-FDA-1088 y también al centro de llamadas REMS al 1-888-423-5436.
REVLIMID es un análogo de la talidomida. La talidomida es un teratógeno humano, que induce una alta frecuencia de defectos congénitos graves y potencialmente mortales, como la amelía (ausencia de extremidades), la focomelia (extremidades cortas), la hipoplasia ósea, la ausencia de huesos, anomalías del oído externo (incluyendo anotia, micropinna, canales auditivos externos pequeños o ausentes), parálisis facial, anomalías oculares (anofthalmos, microphthalmos) y defectos cardíacos congénitos. También se han documentado malformaciones del tracto alimenticio, urinario y genital, y se ha informado de una mortalidad en el momento del parto o poco después en aproximadamente 40% de los lactantes.
La lenalidomida provocó defectos en las extremidades de tipo talidomida en las crías de monos. La lenalidomida cruzó la placenta después de la administración a conejas embarazadas y ratas embarazadas [ver Datos]. Si se usa este fármaco durante el embarazo, o si la paciente queda embarazada mientras toma este fármaco, se debe informar a la paciente del riesgo potencial para el feto.
Si ocurre un embarazo durante el tratamiento, suspender el fármaco inmediatamente. En estas condiciones, remitir a la paciente a un obstetra/ginecólogo experimentado en toxicidad reproductiva para una evaluación y asesoramiento adicionales. Informe cualquier sospecha de exposición fetal a REVLIMID a la FDA a través del programa MedWatch al 1-800-FDA-1088 y también al centro de llamadas REMS al 1-888-423-5436.
El riesgo estimado de fondo de defectos congénitos mayores y abortos espontáneos en la población indicada es desconocido. Todos los embarazos tienen un riesgo de fondo de defectos congénitos, pérdidas o otros resultados adversos. El riesgo estimado de fondo en la población general estadounidense de defectos congénitos mayores es del 2%-4% y de abortos espontáneos es del 15%-20% de los embarazos clínicamente reconocidos.
Datos
Datos de animales
En un estudio de toxicidad del desarrollo embrión-fetal en monos, ocurrieron teratogenicidad, incluyendo defectos en las extremidades similares a los de la talidomida, en las crías cuando las monas embarazadas recibieron lenalidomida oral durante la organogénesis. La exposición (AUC) en monos con la dosis más baja fue 0,17 veces la exposición humana en la dosis humana máxima recomendada (MRHD) de 25 mg. Estudios similares en conejas embarazadas y ratas a 20 veces y 200 veces la MRHD respectivamente, produjeron letalidad embrionaria en conejas y ningún efecto reproductivo adverso en ratas.
En un estudio de desarrollo pre y postnatal en ratas, los animales recibieron lenalidomida desde la organogénesis hasta la lactancia. El estudio reveló algunos efectos adversos en las crías de ratas hembras tratadas con lenalidomida en dosis de hasta 500 mg / kg (aproximadamente 200 veces la dosis humana de 25 mg basada en el área de superficie corporal). Los descendientes masculinos exhibieron un ligero retraso en la maduración sexual y las crías femeninas tuvieron unos pequeños aumentos de peso corporal inferiores durante la gestación cuando se cruzaron con descendientes masculinos. Al igual que con la talidomida, el modelo de ratas puede no abordar adecuadamente todo el espectro de efectos de desarrollo embrión-fetal humanos potenciales de la lenalidomida.
Después de la administración oral diaria de lenalidomida desde el Día 7 de gestación hasta el Día 20 en conejas embarazadas, las concentraciones de lenalidomida en el plasma fetal fueron aproximadamente del 20-40% del Cmáx. materno. Después de una dosis oral única a ratas embarazadas, se detectó lenalidomida en el plasma fetal y los tejidos; las concentraciones de radioactividad en los tejidos fetales fueron generalmente inferiores a las de los tejidos maternos. Estos datos indicaron que la lenalidomida cruzó la placenta.
8.2 Lactancia
Resumen del riesgo
No hay información sobre la presencia de lenalidomida en la leche humana, los efectos de REVLIMID en el niño lactante o los efectos de REVLIMID en la producción de leche. Debido a que muchos fármacos se excretan en la leche humana y debido al potencial de reacciones adversas en los niños lactantes por REVLIMID, se aconseja a las mujeres que no amamanten durante el tratamiento con REVLIMID.
8.3 Mujeres y hombres con potencial reproductivo
Pruebas de embarazo
REVLIMID puede causar daño fetal cuando se administra durante el embarazo [ver Uso en poblaciones específicas (8.1)]. Verificar el estado de embarazo de las Mujeres con potencial reproductivo antes de iniciar la terapia con REVLIMID y durante el tratamiento. Aconsejar a las mujeres con potencial reproductivo que deben evitar el embarazo 4 semanas antes del tratamiento, mientras toman REVLIMID, durante interrupciones de la dosis y durante al menos 4 semanas después de completar el tratamiento.
Las mujeres con potencial reproductivo deben tener 2 pruebas de embarazo negativas antes de iniciarse el tratamiento con REVLIMID. La primera prueba debe realizarse dentro de 10-14 días y la segunda prueba dentro de 24 horas antes de prescribir REVLIMID. Una vez que ha comenzado el tratamiento y durante interrupciones de la dosis, las pruebas de embarazo para mujeres con potencial reproductivo deben realizarse semanalmente durante las primeras 4 semanas de uso, luego las pruebas de embarazo deben repetirse cada 4 semanas en mujeres con ciclos menstruales regulares. Si los ciclos menstruales son irregulares, las pruebas de embarazo deben realizarse cada 2 semanas. Deben realizarse pruebas de embarazo y asesoramiento si una paciente pierde su período o si hay alguna anormalidad en su sangrado menstrual. El tratamiento con REVLIMID debe suspenderse durante esta evaluación.
Anticoncepción
Mujeres
Las mujeres en edad fértil deben comprometerse a abstenerse continuamente de las relaciones sexuales heterosexuales o a utilizar 2 métodos de control de la natalidad fiables simultáneamente: una forma de anticoncepción altamente eficaz – ligadura de trompas, DIU, hormonal (píldoras anticonceptivas, inyecciones, parches hormonales, anillos vaginales o implantes), o vasectomía de la pareja, y 1 método anticonceptivo eficaz adicional – condón masculino de látex o sintético, diafragma o capuchón cervical. La anticoncepción debe comenzar 4 semanas antes de iniciar el tratamiento con REVLIMID, durante la terapia, durante las interrupciones de la dosis y continuar durante 4 semanas después de la interrupción del tratamiento con REVLIMID. La anticoncepción fiable está indicada incluso cuando ha habido antecedentes de infertilidad, a menos que se deba a una histerectomía. Las mujeres en edad fértil deben ser derivadas a un proveedor cualificado de métodos anticonceptivos, si es necesario.
Hombres
La lenalidomida está presente en el semen de los hombres que toman REVLIMID. Por lo tanto, los hombres deben utilizar siempre un condón de látex o sintético durante cualquier contacto sexual con mujeres en edad fértil mientras toman REVLIMID y hasta 4 semanas después de dejar de tomar REVLIMID, incluso si se han sometido a una vasectomía exitosa. Los pacientes de sexo masculino que toman REVLIMID no deben donar esperma y hasta 4 semanas después de dejar de tomar REVLIMID.
8.4 Uso pediátrico
La seguridad y la eficacia no se han establecido en pacientes pediátricos.
8.5 Uso geriátrico
MM en combinación: En general, de los 1613 pacientes del estudio NDMM que recibieron el tratamiento del estudio, el 94% (1521/1613) tenía 65 años de edad o más, mientras que el 35% (561/1613) tenía más de 75 años de edad. El porcentaje de pacientes mayores de 75 años fue similar entre los brazos del estudio (Rd Continuo: 33%; Rd18: 34%; MPT: 33%). En general, en todos los brazos de tratamiento, la frecuencia en la mayoría de las categorías de reacciones adversas (por ejemplo, todas las reacciones adversas, reacciones adversas de grado 3/4, reacciones adversas graves) fue mayor en los sujetos de mayor edad (> 75 años de edad) que en los sujetos más jóvenes (≤ 75 años de edad). Las reacciones adversas de grado 3 o 4 en el sistema corporal de Trastornos generales y condiciones del lugar de administración se informaron de forma constante con una frecuencia mayor (con una diferencia de al menos el 5%) en los sujetos de mayor edad que en los sujetos más jóvenes en todos los brazos de tratamiento. Las reacciones adversas de grado 3 o 4 en los sistemas corporales de Infecciones e Infestaciones, Trastornos cardíacos (incluida la insuficiencia cardíaca y la insuficiencia cardíaca congestiva), Trastornos de la piel y del tejido subcutáneo, y Trastornos renales y urinarios (incluida la insuficiencia renal) también se informaron de forma ligeramente, pero constante, más frecuente (< 5% de diferencia), en los sujetos de mayor edad que en los sujetos más jóvenes en todos los brazos de tratamiento. Para otros sistemas corporales (por ejemplo, Trastornos de la sangre y del sistema linfático, Infecciones e Infestaciones, Trastornos cardíacos, Trastornos vasculares), hubo una tendencia menos consistente para una mayor frecuencia de reacciones adversas de grado 3/4 en los sujetos de mayor edad frente a los sujetos más jóvenes en todos los brazos de tratamiento. Las reacciones adversas graves se informaron generalmente con una frecuencia mayor en los sujetos de mayor edad que en los sujetos más jóvenes en todos los brazos de tratamiento.
Terapia de mantenimiento de MM: En general, el 10% (106/1018) de los pacientes tenía 65 años de edad o más, mientras que ningún paciente tenía más de 75 años de edad. Las reacciones adversas de grado 3 o 4 fueron mayores en el brazo de REVLIMID (más del 5% más) en los pacientes de 65 años de edad o más en comparación con los pacientes más jóvenes. La frecuencia de las reacciones adversas de grado 3 o 4 en los Trastornos de la sangre y del sistema linfático fue mayor en el brazo de REVLIMID (más del 5% más) en los pacientes de 65 años de edad o más en comparación con los pacientes más jóvenes. No hubo un número suficiente de pacientes de 65 años de edad o más en los estudios de mantenimiento de REVLIMID que experimentaron una reacción adversa grave, o que interrumpieron la terapia debido a una reacción adversa, para determinar si los pacientes de edad avanzada responden en relación con la seguridad de forma diferente a los pacientes más jóvenes.
MM después de al menos una terapia previa: De los 703 pacientes con MM que recibieron el tratamiento del estudio en los estudios 1 y 2, el 45% tenía 65 años o más, mientras que el 12% de los pacientes tenía 75 años o más. El porcentaje de pacientes de 65 años o más no fue significativamente diferente entre los grupos de REVLIMID/dexametasona y placebo/dexametasona. De los 353 pacientes que recibieron REVLIMID/dexametasona, el 46% tenía 65 años o más. En ambos estudios, los pacientes > 65 años de edad tenían más probabilidades que los pacientes ≤ 65 años de edad de experimentar TVP, embolia pulmonar, fibrilación auricular e insuficiencia renal después del uso de REVLIMID. No se observaron diferencias en la eficacia entre los pacientes mayores de 65 años de edad y los pacientes más jóvenes.
De los 148 pacientes con del 5q MDS inscritos en el estudio principal, el 38% tenía 65 años o más, mientras que el 33% tenía 75 años o más. Aunque la frecuencia general de reacciones adversas (100%) fue la misma en los pacientes mayores de 65 años de edad que en los pacientes más jóvenes, la frecuencia de reacciones adversas graves fue mayor en los pacientes mayores de 65 años de edad que en los pacientes más jóvenes (54% frente al 33%). Una mayor proporción de pacientes mayores de 65 años de edad abandonaron los estudios clínicos debido a reacciones adversas que la proporción de pacientes más jóvenes (27% frente al 16%). No se observaron diferencias en la eficacia entre los pacientes mayores de 65 años de edad y los pacientes más jóvenes.
De los 134 pacientes con MCL inscritos en el ensayo de MCL, el 63% tenía 65 años o más, mientras que el 22% de los pacientes tenía 75 años o más. La frecuencia general de reacciones adversas fue similar en los pacientes mayores de 65 años de edad y en los pacientes más jóvenes (98% frente al 100%). La incidencia general de reacciones adversas de grado 3 y 4 también fue similar en estos 2 grupos de pacientes (79% frente al 78%, respectivamente). La frecuencia de reacciones adversas graves fue mayor en los pacientes mayores de 65 años de edad que en los pacientes más jóvenes (55% frente al 41%). No se observaron diferencias en la eficacia entre los pacientes mayores de 65 años de edad y los pacientes más jóvenes.
FL o MZL en combinación: En general, el 48% (282/590) de los pacientes tenían 65 años o más, mientras que el 14% (82/590) de los pacientes tenían más de 75 años. La frecuencia general de reacciones adversas fue similar en pacientes de 65 años o más y pacientes más jóvenes para ambos estudios combinados (98%). Las reacciones adversas de grado 3 o 4 fueron más altas en el brazo de REVLIMID (más del 5% más altas) en los pacientes de 65 años o más en comparación con los pacientes más jóvenes (71% frente a 59%). La frecuencia de reacciones adversas de grado 3 o 4 fue más alta en el brazo de REVLIMID (más del 5% más alta) en los pacientes de 65 años o más en comparación con los pacientes más jóvenes en los trastornos de la sangre y el sistema linfático (47% frente a 40%) e infecciones e infestaciones (16% frente a 11%). Las reacciones adversas graves fueron más altas en el brazo de REVLIMID (más del 5% más altas) en los pacientes de 65 años o más en comparación con los pacientes más jóvenes (37% frente a 18%). La frecuencia de reacciones adversas graves fue más alta en el brazo de REVLIMID (más del 5% más alta) en los pacientes de 65 años o más en comparación con los pacientes más jóvenes en infecciones e infestaciones (15% frente a 6%).
Dado que los pacientes de edad avanzada tienen más probabilidades de tener una función renal disminuida, se debe tener cuidado en la selección de la dosis. Monitorear la función renal.
8.6 Insuficiencia renal
Ajuste la dosis inicial de REVLIMID en función del valor de aclaramiento de creatinina y para pacientes en diálisis [ver Dosificación y administración (2.6)].
10 SOBREDOSIS
No hay experiencia específica en el manejo de la sobredosis de REVLIMID en pacientes con MM, MDS, MCL, FL o MZL. En estudios de rango de dosis en sujetos sanos, algunos fueron expuestos a hasta 200 mg (administrados 100 mg BID) y en estudios de dosis única, algunos sujetos fueron expuestos a hasta 400 mg. Prurito, urticaria, erupción cutánea y elevación de las transaminasas hepáticas fueron los principales AE informados. En los ensayos clínicos, la toxicidad limitante de la dosis fue la neutropenia y la trombocitopenia.
11 DESCRIPCIÓN
REVLIMID, un análogo de la talidomida, es un agente inmunomodulador con propiedades antiangiogénicas y antineoplásicas. El nombre químico es 3-(4-amino-1-oxo 1,3-dihidro-2H-isoindol-2-il) piperidina-2,6-diona y tiene la siguiente estructura química:
La fórmula empírica de la lenalidomida es C13H13N3O3, y el peso molecular en gramos es de 259,3.
La lenalidomida es un polvo sólido de color blanquecino a amarillo pálido. Es soluble en mezclas de solventes orgánicos/agua y en solventes acuosos tamponados. La lenalidomida es más soluble en solventes orgánicos y en soluciones con pH bajo. La solubilidad fue significativamente menor en tampones menos ácidos, oscilando entre 0,4 y 0,5 mg/ml aproximadamente. La lenalidomida tiene un átomo de carbono asimétrico y puede existir en las formas ópticamente activas S(-) y R(+), y se produce como una mezcla racémica con una rotación óptica neta de cero.
REVLIMID está disponible en cápsulas de 2,5 mg, 5 mg, 10 mg, 15 mg, 20 mg y 25 mg para administración oral. Cada cápsula contiene lenalidomida como ingrediente activo y los siguientes ingredientes inactivos: lactosa anhidra, celulosa microcristalina, croscarmelosa sódica y estearato de magnesio. La cubierta de la cápsula de 5 mg y 25 mg contiene gelatina, dióxido de titanio y tinta negra. La cubierta de la cápsula de 2,5 mg y 10 mg contiene gelatina, FD&C azul n.° 2, óxido de hierro amarillo, dióxido de titanio y tinta negra. La cubierta de la cápsula de 15 mg contiene gelatina, FD&C azul n.° 2, dióxido de titanio y tinta negra. La cubierta de la cápsula de 20 mg contiene gelatina, FD&C azul n.° 2, óxido de hierro amarillo, dióxido de titanio y tinta negra.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
La lenalidomida es un análogo de la talidomida con propiedades inmunomoduladoras, antiangiogénicas y antineoplásicas. Las actividades celulares de la lenalidomida están mediadas por su diana cereblon, un componente de un complejo enzimático de ubiquitina ligasa E3 de anillo de cullin. In vitro, en presencia del fármaco, las proteínas sustrato (incluidas Aiolos, Ikaros y CK1α) se dirigen a la ubiquitinación y posterior degradación, lo que provoca efectos citotóxicos e inmunomoduladores directos. La lenalidomida inhibe la proliferación e induce la apoptosis de determinadas células tumorales hematopoyéticas, como el MM, el linfoma de células del manto y los síndromes mielodisplásicos del (5q), el linfoma folicular y el linfoma de la zona marginal in vitro. La lenalidomida provoca un retraso en el crecimiento tumoral en algunos modelos tumorales hematopoyéticos no clínicos in vivo, incluido el MM. Las propiedades inmunomoduladoras de la lenalidomida incluyen el aumento del número y la activación de las células T y las células asesinas naturales (NK), lo que conduce a una citotoxicidad celular dependiente de anticuerpos (ADCC) directa y mejorada a través del aumento de la secreción de interleucina-2 e interferón-gamma, el aumento del número de células NKT y la inhibición de las citocinas proinflamatorias (por ejemplo, TNF-α e IL-6) por parte de los monocitos. En las células MM, la combinación de lenalidomida y dexametasona sinergiza la inhibición de la proliferación celular y la inducción de la apoptosis. La combinación de lenalidomida y rituximab aumenta la ADCC y la apoptosis tumoral directa en las células del linfoma folicular y aumenta la ADCC en las células del linfoma de la zona marginal en comparación con rituximab solo in vitro.
12.2 Farmacodinámica
Electrofisiología cardíaca
El efecto de la lenalidomida sobre el intervalo QTc se evaluó en 60 sujetos varones sanos en un estudio exhaustivo de QT. A una dosis dos veces superior a la máxima recomendada, la lenalidomida no prolongó el intervalo QTc. El límite superior más amplio del IC del 90 % bilateral para las diferencias de medias entre lenalidomida y placebo fue inferior a 10 ms.
12.3 Farmacocinética
Absorción
Tras la administración de dosis únicas y múltiples de REVLIMID en pacientes con MM o SMD, las concentraciones plasmáticas máximas se alcanzaron entre 0,5 y 6 horas después de la dosis. La disposición farmacocinética de dosis únicas y múltiples de lenalidomida es lineal, y los valores de AUC y Cmáx aumentan proporcionalmente con la dosis. La administración de dosis múltiples de REVLIMID a la dosis recomendada no da lugar a la acumulación del fármaco.
La administración de una dosis única de 25 mg de REVLIMID con una comida rica en grasas en sujetos sanos reduce el grado de absorción, con una disminución aproximada del 20 % en el AUC y del 50 % en la Cmáx. En los ensayos en los que se estableció la eficacia y la seguridad de REVLIMID, el fármaco se administró independientemente de la ingesta de alimentos. REVLIMID puede administrarse con o sin alimentos.
La tasa de absorción oral de la lenalidomida en pacientes con MCL es similar a la observada en pacientes con MM o SMD.
Distribución
In vitro, la unión de [14C]-lenalidomida a las proteínas plasmáticas es de aproximadamente el 30 %.
La lenalidomida está presente en el semen a las 2 horas (1379 ng/eyaculado) y 24 horas (35 ng/eyaculado) después de la administración de 25 mg de REVLIMID al día.
Eliminación
La semivida media de la lenalidomida es de 3 horas en sujetos sanos y de 3 a 5 horas en pacientes con MM, SMD o MCL.
Metabolismo
La lenalidomida experimenta un metabolismo limitado. La lenalidomida inalterada es el componente circulante predominante en los seres humanos. Se han identificado dos metabolitos: 5-hidroxi-lenalidomida y N-acetil-lenalidomida; cada uno de ellos constituye menos del 5 % de los niveles del fármaco original en circulación.
Excreción
La eliminación es principalmente renal. Tras una única administración oral de 25 mg de [14C]-lenalidomida a sujetos sanos, aproximadamente el 90 % y el 4 % de la dosis radiactiva se eliminó en la orina y las heces, respectivamente, en un plazo de diez días. Aproximadamente el 82 % de la dosis radiactiva se excretó como lenalidomida en la orina en las 24 horas siguientes. La hidroxi-lenalidomida y la N-acetil-lenalidomida representaron el 4,6 % y el 1,8 % de la dosis excretada, respectivamente. El aclaramiento renal de la lenalidomida supera la tasa de filtración glomerular.
Poblaciones específicas
Insuficiencia renal: Ocho sujetos con insuficiencia renal leve (aclaramiento de creatinina (CLcr) de 50 a 79 ml/min calculado mediante la fórmula de Cockcroft-Gault), 9 sujetos con insuficiencia renal moderada (CLcr de 30 a 49 ml/min), 4 sujetos con insuficiencia renal grave (CLcr < 30 ml/min) y 6 pacientes con enfermedad renal terminal (ERT) que requerían diálisis recibieron una dosis única de 25 mg de REVLIMID. Tres sujetos sanos de edad similar con función renal normal (CLcr > 80 ml/min) también recibieron una dosis única de 25 mg de REVLIMID. A medida que el CLcr disminuía, la semivida aumentaba y el aclaramiento del fármaco disminuía linealmente. Los pacientes con insuficiencia moderada y grave presentaron un aumento de 3 veces en la semivida y una disminución del 66 % al 75 % en el aclaramiento del fármaco en comparación con los sujetos sanos. Los pacientes en hemodiálisis (n = 6) presentaron un aumento aproximado de 4,5 veces en la semivida y una disminución del 80 % en el aclaramiento del fármaco en comparación con los sujetos sanos. Aproximadamente el 30 % del fármaco en el organismo se eliminó durante una sesión de hemodiálisis de 4 horas.
Ajuste la dosis inicial de REVLIMID en pacientes con insuficiencia renal según el valor de CLcr [consulte Dosificación y administración (2.6)].
Insuficiencia hepática: La insuficiencia hepática leve (definida como bilirrubina total > 1 a 1,5 veces el límite superior normal (LSN) o cualquier aspartato transaminasa superior al LSN) no influyó en la disposición de lenalidomida. No se dispone de datos farmacocinéticos para pacientes con insuficiencia hepática moderada a grave.
Otros factores intrínsecos: La edad (39 a 85 años), el peso corporal (33 a 135 kg), el sexo, la raza y el tipo de neoplasias hematológicas (MM, SMD o MCL) no tuvieron un efecto clínicamente relevante sobre la depuración de lenalidomida en pacientes adultos.
Interacciones medicamentosas
La administración conjunta de una dosis única o de múltiples dosis de dexametasona (40 mg) no tuvo un efecto clínicamente relevante sobre la farmacocinética de dosis múltiples de REVLIMID (25 mg).
La administración conjunta de REVLIMID (25 mg) después de múltiples dosis de un inhibidor de la P-gp como la quinidina (600 mg dos veces al día) no aumentó significativamente la Cmáx ni el AUC de lenalidomida.
La administración conjunta del inhibidor y sustrato de la P-gp temsirolimus (25 mg) con REVLIMID (25 mg) no alteró significativamente la farmacocinética de lenalidomida, temsirolimus o sirolimus (metabolito de temsirolimus).
Los estudios in vitro demostraron que REVLIMID es un sustrato de la glicoproteína P (P-gp). REVLIMID no es un sustrato de la proteína de resistencia al cáncer de mama humano (BCRP), los transportadores de la proteína de resistencia a múltiples fármacos (MRP) MRP1, MRP2 o MRP3, los transportadores de aniones orgánicos (OAT) OAT1 y OAT3, el polipéptido transportador de aniones orgánicos 1B1 (OATP1B1), los transportadores de cationes orgánicos (OCT) OCT1 y OCT2, la proteína de extrusión de múltiples fármacos y toxinas (MATE) MATE1 y los nuevos transportadores de cationes orgánicos (OCTN) OCTN1 y OCTN2. La lenalidomida no es un inhibidor de la P-gp, la bomba de exportación de sales biliares (BSEP), la BCRP, la MRP2, la OAT1, la OAT3, la OATP1B1, la OATP1B3 ni la OCT2. La lenalidomida no inhibe ni induce las isoenzimas del CYP450. Además, la lenalidomida no inhibe la formación de glucuronidación de la bilirrubina en los microsomas hepáticos humanos con UGT1A1 genotipado como UGT1A1*1/*1, UGT1A1*1/*28 y UGT1A1*28/*28.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
No se han realizado estudios de carcinogenicidad con lenalidomida.
Lenalidomida no fue mutagénica en la prueba de mutación inversa bacteriana (prueba de Ames) y no indujo aberraciones cromosómicas en linfocitos de sangre periférica humana cultivados, ni mutaciones en el locus de la timidina quinasa (tk) de las células de linfoma de ratón L5178Y. Lenalidomida no aumentó la transformación morfológica en la prueba de embrión de hámster sirio ni indujo micronúcleos en los eritrocitos policromáticos de la médula ósea de ratas macho.
Un estudio de fertilidad y desarrollo embrionario temprano en ratas, con administración de lenalidomida hasta 500 mg/kg (aproximadamente 200 veces la dosis humana de 25 mg, basada en el área de superficie corporal) no produjo toxicidad parental ni efectos adversos sobre la fertilidad.
14 ESTUDIOS CLÍNICOS
14.1 Mieloma múltiple
Ensayo clínico abierto, aleatorizado, en pacientes con MM de diagnóstico reciente:
Se realizó un ensayo de 3 grupos, abierto, aleatorizado, multicéntrico, con 1623 pacientes para comparar la eficacia y seguridad de REVLIMID y dosis bajas de dexametasona (Rd) administradas durante 2 duraciones diferentes con la de melfalán, prednisona y talidomida (MPT) en pacientes con MM de diagnóstico reciente que no eran candidatos a trasplante de células madre. En el primer grupo del estudio, se administró Rd de forma continua hasta la progresión de la enfermedad [Grupo Rd Continuo]. En el segundo grupo, se administró Rd durante un máximo de dieciocho ciclos de 28 días [72 semanas, Grupo Rd18]). En el tercer grupo, se administró melfalán, prednisona y talidomida (MPT) durante un máximo de doce ciclos de 42 días (72 semanas). A efectos de este estudio, un paciente menor de 65 años no era candidato a TCT si se negaba a someterse a la terapia TCT o si no tenía acceso a la TCT por motivos económicos u otros. Los pacientes se estratificaron en la aleatorización por edad (≤75 frente a >75 años), estadio (estadios ISS I y II frente a estadio III) y país.
Los pacientes de los grupos Rd Continuo y Rd18 recibieron 25 mg de REVLIMID una vez al día los días 1 a 21 de ciclos de 28 días. La dexametasona se dosificó a 40 mg una vez al día los días 1, 8, 15 y 22 de cada ciclo de 28 días. En los pacientes mayores de 75 años, la dosis inicial de dexametasona fue de 20 mg por vía oral una vez al día los días 1, 8, 15 y 22 de ciclos repetidos de 28 días. La dosis inicial y las pautas para Rd Continuo y Rd18 se ajustaron en función de la edad y la función renal. Todos los pacientes recibieron anticoagulación profiláctica, siendo la más utilizada la aspirina.
Las características demográficas y basales relacionadas con la enfermedad de los pacientes estaban equilibradas entre los 3 grupos. En general, los sujetos del estudio presentaban una enfermedad en estadio avanzado. De la población total del estudio, la mediana de edad fue de 73 años en los 3 grupos, con un 35 % de pacientes mayores de 75 años; el 59 % presentaba estadio ISS I/II; el 41 % presentaba estadio ISS III; el 9 % presentaba insuficiencia renal grave (aclaramiento de creatinina [CLcr] < 30 ml/min); el 23 % presentaba insuficiencia renal moderada (CLcr > 30 a 50 ml/min; el 44 % presentaba insuficiencia renal leve (CLcr > 50 a 80 ml/min). En cuanto al estado funcional ECOG, el 29 % eran de grado 0, el 49 % de grado 1, el 21 % de grado 2 y el 0,4 % de grado ≥ 3.
La variable principal de eficacia, la supervivencia libre de progresión (SLP), se definió como el tiempo transcurrido desde la aleatorización hasta la primera documentación de progresión de la enfermedad según la determinación del Comité Independiente de Adjudicación de Respuesta (CAIR), según los criterios del International Myeloma Working Group [IMWG] o la muerte por cualquier causa, lo que ocurriera primero durante el estudio hasta el final de la fase de seguimiento de la SLP. Para el análisis de eficacia de todas las variables, la comparación principal se realizó entre los grupos Rd Continuo y MPT. Los resultados de eficacia se resumen en la siguiente tabla. La SLP fue significativamente mayor con Rd Continuo que con MPT: HR 0,72 (IC del 95 %: 0,61-0,85 p <0,0001). Un porcentaje menor de sujetos en el grupo Rd Continuo en comparación con el grupo MPT tuvo eventos de SLP (52 % frente a 61 %, respectivamente). La mejora en la mediana del tiempo de SLP en el grupo Rd Continuo en comparación con el grupo MPT fue de 4,3 meses. La tasa de respuesta del mieloma fue mayor con Rd Continuo en comparación con MPT (75,1 % frente a 62,3 %), con una respuesta completa en el 15,1 % de los pacientes del grupo Rd Continuo frente al 9,3 % en el grupo MPT. La mediana del tiempo hasta la primera respuesta fue de 1,8 meses en el grupo Rd Continuo frente a 2,8 meses en el grupo MPT.
Para el análisis provisional de la SG con datos de corte del 3 de marzo de 2014, la mediana del tiempo de seguimiento de todos los pacientes supervivientes es de 45,5 meses, con 697 eventos de muerte, que representan el 78 % de los eventos preespecificados necesarios para el análisis final previsto de la SG (697/896 de los eventos finales de la SG). La HR de la SG observada fue de 0,75 para Rd Continuo frente a MPT (IC del 95 % = 0,62, 0,90).
Tabla 13: Resumen de los resultados de eficacia – Estudio MM-020 (Población por intención de tratar)
RC = respuesta completa; d = dosis baja de dexametasona; HR = hazard ratio (cociente de riesgos instantáneos); CAIR = Comité Independiente de Adjudicación de Respuesta; M = melfalán; NE = no estimable; SG = supervivencia global; P = prednisona; SLP = supervivencia libre de progresión; RP = respuesta parcial; R = REVLIMID; Rd Continuo = Rd administrado hasta la documentación de progresión de la enfermedad; Rd18 = Rd administrado durante ≤ 18 ciclos; T = talidomida; MRPB = muy buena respuesta parcial; vs = versus. a La mediana se basa en la estimación de Kaplan-Meier. b El intervalo de confianza (IC) del 95 % sobre la mediana. c Basado en el modelo de riesgos proporcionales de Cox que compara las funciones de riesgo asociadas a los grupos de tratamiento indicados. d El valor p se basa en la prueba de log-rank no estratificada de las diferencias de la curva de Kaplan-Meier entre los grupos de tratamiento indicados. e Mejor evaluación de la respuesta durante la fase de tratamiento del estudio. f Incluyendo a los pacientes sin datos de evaluación de la respuesta o cuya única evaluación fue “respuesta no evaluable”. g Fecha de corte de datos = 24 de mayo de 2013. h Fecha de corte de datos = 3 de marzo de 2014.
Rd Continuo
(N = 535)
Rd18
(N = 541)
MPT
(N = 547)
SLP – CAIR (meses)g
Número de eventos de SLP
278 (52)
348 (64.3)
334 (61.1)
Medianaa de tiempo hasta la PFS, meses (IC del 95%)b
25.5 (20.7, 29.4)
20.7 (19.4, 22)
21.2 (19.3, 23.2)
HR [IC del 95%]c; valor de pd
Rd Continuo frente a MPT
0.72 (0.61, 0.85); <0.0001
Rd Continuo frente a Rd18
0.70 (0.60, 0.82)
Rd18 frente a MPT
1.03 (0.89, 1.20)
Supervivencia global (meses)h
Número de eventos de muerte
208 (38.9)
228 (42.1)
261 (47.7)
Medianaa de tiempo hasta la SG, meses (IC del 95%)b
58.9 (56, NE)f
56.7 (50.1, NE)
48.5 (44.2, 52 )
HR [IC del 95%]c
Rd Continuo frente a MPT
0.75 (0.62, 0.90)
Rd Continuo frente a Rd18
0.91 (0.75, 1.09)
Rd18 frente a MPT
0.83 (0.69, 0.99)
Tasa de respuestae – IRAC, n (%)g
CR
81 (15.1)
77 (14.2)
51 (9.3)
VGPR
152 (28.4)
154 (28.5)
103 (18.8)
PR
169 (31.6)
166 (30.7)
187 (34.2)
Respuesta global: CR, VGPR o PR
402 (75.1)
397 (73.4)
341 (62.3)
Curvas de Kaplan-Meier de Supervivencia Libre de Progresión Basadas en la Evaluación IRAC (Población ITT MM) Entre los Grupos Rd Continuo, Rd18 y MPT Fecha límite: 24 de mayo de 2013
CI = intervalo de confianza; d = dexametasona en dosis baja; HR = ratio de riesgo; IRAC = Comité Independiente de Adjudicación de Respuesta; M = melfalan; P = prednisona; R = REVLIMID; Rd Continuo = Rd administrado hasta la documentación de enfermedad progresiva; Rd18 = Rd administrado por ≤ 18 ciclos; T = talidomida.
Curvas de Kaplan-Meier de Supervivencia Global (Población ITT MM) Entre los Grupos Rd Continuo, Rd18 y MPT Fecha límite: 03 de marzo de 2014
CI = intervalo de confianza; d = dexametasona en dosis baja; HR = ratio de riesgo; M = melfalan; P = prednisona; R = REVLIMID; Rd Continuo = Rd administrado hasta la documentación de enfermedad progresiva; Rd18 = Rd administrado por ≤18 ciclos; T = talidomida.
Estudios Clínicos Aleatorizados, Controlados con Placebo – Mantenimiento Tras Auto-HSCT:
Se realizaron dos estudios multicéntricos, aleatorizados, doble ciego, en grupos paralelos, controlados con placebo para evaluar la eficacia y seguridad de la terapia de mantenimiento con REVLIMID en el tratamiento de pacientes con MM después de auto-HSCT. En el Estudio de Mantenimiento 1, eran elegibles los pacientes de entre 18 y 70 años que hubieran recibido terapia de inducción seguida de auto-HSCT. La terapia de inducción debía haber tenido lugar dentro de 12 meses. Entre 90 y 100 días después de la auto-HSCT, los pacientes con al menos una respuesta de enfermedad estable fueron aleatorizados 1:1 para recibir mantenimiento con REVLIMID o placebo. En el Estudio de Mantenimiento 2, eran elegibles los pacientes de < 65 años en el momento del diagnóstico que hubieran recibido terapia de inducción seguida de auto-HSCT y hubieran alcanzado al menos una respuesta de enfermedad estable en el momento de la recuperación hematológica. Dentro de los 6 meses después de la auto-HSCT, los pacientes fueron aleatorizados 1:1 para recibir mantenimiento con REVLIMID o placebo. Los pacientes elegibles para ambos ensayos debían tener CLcr ≥ 30 mL/minuto.
En ambos estudios, la dosis de mantenimiento con REVLIMID era de 10 mg una vez al día en los días 1-28 de ciclos repetidos de 28 días, podía aumentarse a 15 mg una vez al día después de 3 meses en ausencia de toxicidad limitante de dosis, y el tratamiento debía continuar hasta la progresión de la enfermedad o la retirada del paciente por otra razón. La dosis se reducía, o el tratamiento se interrumpía temporalmente o se detenía, según fuera necesario para gestionar la toxicidad. Un aumento de dosis a 15 mg una vez al día ocurrió en 135 pacientes (58%) en el Estudio de Mantenimiento 1, y en 185 pacientes (60%) en el Estudio de Mantenimiento 2.
Las características demográficas y basales relacionadas con la enfermedad de los pacientes eran similares en los dos estudios y reflejaban una población típica de MM después de auto-HSCT (ver Tabla 14).
Tabla 14: Características Basales Demográficas y Relacionadas con la Enfermedad – Estudios de Mantenimiento MM 1 y 2
Fecha límite de datos = 1 de marzo de 2015.
Estudio de Mantenimiento 1
Estudio de Mantenimiento 2
REVLIMID
N = 231
Placebo
N = 229
REVLIMID
N = 307
Placebo
N = 307
Edad (años)
Mediana
58
58
57.5
58.1
(Min, max)
(29, 71)
(39, 71)
(22.7, 68.3)
(32.3, 67)
Sexo, n (%)
Masculino
121 (52)
129 (56)
169 (55)
181 (59)
Femenino
110 (48)
100 (44)
138 (45)
126 (41)
Etapa ISS al Diagnóstico, n (%)
Etapa I o II
120 (52)
131 (57)
232 (76)
250 (81)
Stage I
62 (27)
85 (37)
128 (42)
143 (47)
Stage II
58 (25)
46 (20)
104 (34)
107 (35)
Etapa III
39 (17)
35 (15)
66 (21)
46 (15)
Faltan datos
72 (31)
63 (28)
9 (3)
11 (4)
CrCl en el Post-auto-HSCT, n (%)
< 50 mL/min
23 (10)
16 (7)
10 (3)
9 (3)
≥ 50 mL/min
201 (87)
204 (89)
178 (58)
200 (65)
Faltan datos
7 (3)
9 (4)
119 (39)
98 (32)
El criterio principal de valoración de la eficacia de ambos estudios fue la supervivencia libre de progresión (SLP) definida desde la aleatorización hasta la fecha de progresión o muerte, lo que ocurriera primero; los estudios individuales no tuvieron la potencia suficiente para un criterio de valoración de la supervivencia global. Ambos estudios se desenmascararon por recomendación de sus respectivos comités de monitorización de datos y después de superar los umbrales respectivos para los análisis provisionales planificados de la SLP. Después del desenmascaramiento, se siguió a los pacientes como antes. A los pacientes del grupo placebo del Estudio de mantenimiento 1 se les permitió pasar a recibir REVLIMID antes de la progresión de la enfermedad (76 pacientes [33 %] pasaron a REVLIMID); no se recomendó el paso a los pacientes del Estudio de mantenimiento 2. Los resultados de eficacia se resumen en la siguiente tabla. En ambos estudios, el análisis primario de la SLP en el momento del desenmascaramiento fue significativamente más largo con REVLIMID en comparación con placebo: Estudio de mantenimiento 1 HR 0,38 (IC del 95 %: 0,27-0,54 p < 0,001) y Estudio de mantenimiento 2 HR 0,50 (IC del 95 %: 0,39-0,64 p < 0,001). Para ambos estudios, la SLP se actualizó con una fecha de corte del 1 de marzo de 2015, como se muestra en la tabla y en los siguientes gráficos de Kaplan-Meier. Con un seguimiento más largo (mediana de 72,4 y 86,0 meses, respectivamente), los análisis actualizados de la SLP para ambos estudios siguen mostrando una ventaja de la SLP para REVLIMID en comparación con placebo: Estudio de mantenimiento 1 HR 0,38 (IC del 95 %: 0,28-0,50) con una mediana de la SLP de 68,6 meses y Estudio de mantenimiento 2 HR 0,53 (IC del 95 %: 0,44-0,64) con una mediana de la SLP de 46,3 meses.
En la Tabla 15 se proporciona un análisis descriptivo de los datos de SG con una fecha de corte del 1 de febrero de 2016. El tiempo medio de seguimiento fue de 81,6 y 96,7 meses para el Estudio de mantenimiento 1 y el Estudio de mantenimiento 2, respectivamente. La mediana de la SG fue de 111,0 y 84,2 meses para REVLIMID y placebo, respectivamente, para el Estudio de mantenimiento 1, y de 105,9 y 88,1 meses, para REVLIMID y placebo, respectivamente, para el Estudio de mantenimiento 2.
Tabla 15: Supervivencia libre de progresión y supervivencia global desde la aleatorización en los Estudios de mantenimiento 1 y 2 de MM (Población con intención de tratar posterior al auto-TCMH)
Fecha de desenmascaramiento en el Estudio de mantenimiento 1 y 2 = 17 de diciembre de 2009 y 7 de julio de 2010, respectivamente. Auto-TCMH = trasplante autólogo de células madre hematopoyéticas; IC = intervalo de confianza; ITT = intención de tratar; NE = no estimable; PFS = supervivencia libre de progresión. La SLP en el momento del desenmascaramiento para el Estudio de mantenimiento 2 se basó en la evaluación de un Comité de Revisión Independiente. Todos los demás análisis de la SLP se basaron en la evaluación del investigador. Nota: La mediana se basa en la estimación de Kaplan-Meier, con IC del 95 % sobre el tiempo medio global de la SLP. El índice de riesgo se basa en un modelo de riesgos proporcionales estratificado por factores de estratificación que comparan las funciones de riesgo asociadas a los grupos de tratamiento (REVLIMID:placebo).
Estudio de mantenimiento 1
Estudio de mantenimiento 2
REVLIMID
N = 231
Placebo
N = 229
REVLIMID
N = 307
Placebo
N = 307
SLP en el desenmascaramiento
Eventos de la SLP n (%)
46 (20)
98 (43)
103 (34)
160 (52)
Mediana en meses [IC del 95 %]
33,9 [NE, NE]
19 [16,2, 25,6]
41,2 [38,3, NE]
23,0 [21,2, 28,0]
Índice de riesgo [IC del 95 %]
0,38 [0,27, 0,54]
0,50 [0,39, 0,64]
Valor p de la prueba logarítmica
< 0,001
< 0,001
SLP en el análisis actualizado
1 de marzo de 2015 (Estudios 1 y 2)
Eventos de la SLP n (%)
97 (42)
116 (51)
191 (62)
248 (81)
Mediana en meses [IC del 95 %]
68,6 [52,8, NE]
22,5 [18,8, 30,0]
46,3 [40,1, 56,6]
23,8 [21,0, 27,3]
Hazard Ratio [95% CI]
0.38 [0.28, 0.50]
0.53 [0.44, 0.64]
SG al Actualizar el Análisis
1 de febrero de 2016 (Estudios 1 y 2)
Eventos de SG n (%)
82 (35)
114 (50)
143 (47)
160 (52)
Mediana en meses [95% CI]
111 [101.8, NE]
84.2 [71.0, 102.7]
105.9 [88.8, NE]
88.1 [80.7, 108.4]
Hazard Ratio [95% CI]
0.59 [0.44, 0.78]
0.90 [0.72, 1.13]
Curvas de Kaplan-Meier de supervivencia sin progresión desde la aleatorización (población con intención de tratar después del trasplante autólogo de células madre hematopoyéticas) en el Estudio 1 de mantenimiento de mieloma múltiple entre los grupos de REVLIMID y placebo (fecha de corte actualizada: 1 de marzo de 2015)
Auto-HSCT = trasplante autólogo de células madre hematopoyéticas; IC = intervalo de confianza; CRI = cociente de riesgos instantáneos; ITT = intención de tratar; KM = Kaplan-Meier; SSP = supervivencia sin progresión; vs = versus.
Curvas de Kaplan-Meier de supervivencia sin progresión desde la aleatorización (población con intención de tratar después del trasplante autólogo de células madre hematopoyéticas) en el Estudio 2 de mantenimiento de mieloma múltiple entre los grupos de REVLIMID y placebo (fecha de corte actualizada: 1 de marzo de 2015)
Auto-HSCT = trasplante autólogo de células madre hematopoyéticas; IC = intervalo de confianza; CRI = cociente de riesgos instantáneos; ITT = intención de tratar; KM = Kaplan-Meier; NE = no estimable; SSP = supervivencia sin progresión; vs = versus.
Estudios clínicos aleatorizados, abiertos, en pacientes con mieloma múltiple después de al menos un tratamiento previo
Se realizaron dos estudios aleatorizados (Estudios 1 y 2) para evaluar la eficacia y seguridad de REVLIMID. Estos estudios multicéntricos, multinacionales, doble ciego y controlados con placebo compararon REVLIMID más dosis altas de dexametasona por vía oral con la terapia de dexametasona sola en pacientes con mieloma múltiple que habían recibido al menos un tratamiento previo. En estos estudios se incluyeron pacientes con recuentos absolutos de neutrófilos (RAN) ≥ 1000/mm3, recuentos de plaquetas ≥ 75 000/mm3, creatinina sérica ≤ 2.5 mg/dL, SGOT/AST o SGPT/ALT sérica ≤ 3 x límite superior normal (LSN) y bilirrubina directa sérica ≤ 2 mg/dL.
En ambos estudios, los pacientes del grupo de REVLIMID/dexametasona tomaron 25 mg de REVLIMID por vía oral una vez al día los días 1 a 21 y una cápsula de placebo equivalente una vez al día los días 22 a 28 de cada ciclo de 28 días. Los pacientes del grupo de placebo/dexametasona tomaron 1 cápsula de placebo los días 1 a 28 de cada ciclo de 28 días. Los pacientes de ambos grupos de tratamiento tomaron 40 mg de dexametasona por vía oral una vez al día los días 1 a 4, 9 a 12 y 17 a 20 de cada ciclo de 28 días durante los primeros 4 ciclos de terapia.
La dosis de dexametasona se redujo a 40 mg por vía oral una vez al día los días 1 a 4 de cada ciclo de 28 días después de los primeros 4 ciclos de terapia. En ambos estudios, el tratamiento debía continuar hasta la progresión de la enfermedad.
En ambos estudios, se permitieron ajustes de dosis en función de los hallazgos clínicos y de laboratorio. Se permitieron reducciones de dosis secuenciales a 15 mg diarios, 10 mg diarios y 5 mg diarios por toxicidad [ver Dosis y administración (2.1)].
La Tabla 16 resume las características basales de los pacientes y de la enfermedad en los dos estudios. En ambos estudios, las características basales demográficas y relacionadas con la enfermedad fueron comparables entre los grupos de REVLIMID/dexametasona y placebo/dexametasona.
Tabla 16: Características demográficas y relacionadas con la enfermedad al inicio del estudio: Estudios de mieloma múltiple 1 y 2
Estudio 1
Estudio 2
REVLIMID/Dex
N=177
Placebo/Dex
N=176
REVLIMID/Dex
N=176
Placebo/Dex
N=175
Características del paciente
Edad (años)
Mediana
64
62
63
64
Mín., Máx.
36, 86
37, 85
33, 84
40, 82
Sexo
Masculino
106 (60%)
104 (59%)
104 (59%)
103 (59%)
Femenino
71 (40%)
72 (41%)
72 (41%)
72 (41%)
Raza/etnia
Blanco
141(80%)
148 (84%)
172 (98%)
175 (100%)
Otro
36 (20%)
28 (16%)
4 (2%)
0 (0%)
ECOG Performance
Status 0-1
157 (89%)
168 (95%)
150 (85%)
144 (82%)
Características de la enfermedad
Estadio del mieloma múltiple (Durie-Salmon)
I
3%
3%
6%
5%
II
32%
31%
28%
33%
III
64%
66%
65%
63%
β2-microglobulin (mg/L)
≤ 2.5 mg/L
52 (29%)
51 (29%)
51 (29%)
48 (27%)
> 2.5 mg/L
125 (71%)
125 (71%)
125 (71%)
127 (73%)
Número de terapias previas
1
38%
38%
32%
33%
≥ 2
62%
62%
68%
67%
Tipos de terapias previas
Stem Cell Transplantation
62%
61%
55%
54%
Talidomida
42%
46%
30%
38%
Dexamethasone
81%
71%
66%
69%
Bortezomib
11%
11%
5%
4%
Melphalan
33%
31%
56%
52%
Doxorubicin
55%
51%
56%
57%
El criterio principal de valoración de la eficacia en ambos estudios fue el tiempo hasta la progresión (TTP). El TTP se definió como el tiempo desde la aleatorización hasta la primera aparición de enfermedad progresiva.
Los análisis provisionales planificados de ambos estudios mostraron que la combinación de REVLIMID/dexametasona fue significativamente superior a la dexametasona sola para el TTP. Los estudios se desenmascaron para permitir que los pacientes del grupo de placebo/dexametasona recibieran tratamiento con la combinación de REVLIMID/dexametasona. Para ambos estudios, se analizaron los datos de supervivencia de seguimiento ampliado con cruces. En el estudio 1, la mediana del tiempo de supervivencia fue de 39.4 meses (IC del 95%: 32.9, 47.4) en el grupo de REVLIMID/dexametasona y de 31.6 meses (IC del 95%: 24.1, 40.9) en el grupo de placebo/dexametasona, con una razón de riesgo de 0.79 (IC del 95%: 0.61-1.03). En el estudio 2, la mediana del tiempo de supervivencia fue de 37.5 meses (IC del 95%: 29.9, 46.6) en el grupo de REVLIMID/dexametasona y de 30.8 meses (IC del 95%: 23.5, 40.3) en el grupo de placebo/dexametasona, con una razón de riesgo de 0.86 (IC del 95%: 0.65-1.14).
Tabla 17: Resultados del TTP en el estudio 1 y el estudio 2 del MM
Estudio 1
Estudio 2
REVLIMID/Dex
N=177
Placebo/Dex
N=176
REVLIMID/Dex
N=176
Placebo/Dex
N=175
TTP
Eventos n (%)
73 (41)
120 (68)
68 (39)
130 (74)
Mediana del TTP en meses [IC del 95%]
13.9 [9.5, 18.5]
4.7 [3.7, 4.9]
12.1 [9.5, NE]
4.7 [3.8, 4.8]
Razón de riesgo [IC del 95%]
0.285 [0.210, 0.386]
0.324 [0.240, 0.438]
Valor p de la prueba de log-rank 3
<0.001
<0.001
Respuesta
Respuesta completa (RC) n (%)
23 (13)
1 (1)
27 (15)
7 (4)
Respuesta parcial (RP/RP) n (%)
84 (48)
33 (19)
77 (44)
34 (19)
Respuesta general n (%)
107 (61)
34 (19)
104 (59)
41 (23)
Valor p
<0.001
<0.001
Razón de probabilidad [IC del 95%]
6.38 [3.95, 10.32]
4.72 [2.98, 7.49]
Estimación de Kaplan-Meier del tiempo hasta la progresión: Estudio MM 1
Estimación de Kaplan-Meier del tiempo hasta la progresión: Estudio MM 2
14.2 Síndromes mielodisplásicos (MDS) con anomalía citogenética con deleción 5q
Se evaluó la eficacia y seguridad de REVLIMID en pacientes con anemia dependiente de transfusiones con riesgo bajo o intermedio-1 de MDS con una anomalía citogenética 5q (q31-33) de forma aislada o con anomalías citogenéticas adicionales, a una dosis de 10 mg una vez al día o 10 mg una vez al día durante 21 días cada 28 días en un estudio abierto, de un solo brazo y multicéntrico. El estudio principal no fue diseñado ni tuvo la potencia estadística para comparar de forma prospectiva la eficacia de los 2 regímenes de dosificación. Se permitieron reducciones de dosis secuenciales a 5 mg diarios y 5 mg en días alternos, así como retrasos en la dosis, en caso de toxicidad [Dosificación y administración (2.2)].
En este estudio principal se reclutó a 148 pacientes que tenían anemia dependiente de transfusiones de glóbulos rojos. La dependencia de transfusiones de glóbulos rojos se definió como haber recibido ≥ 2 unidades de glóbulos rojos en las 8 semanas previas al tratamiento del estudio. En el estudio se reclutó a pacientes con recuentos absolutos de neutrófilos (RAN) ≥ 500/mm3, recuentos de plaquetas ≥ 50 000/mm3, creatinina sérica ≤ 2.5 mg/dL, SGOT/AST o SGPT/ALT sérica ≤ 3 veces el límite superior de la normalidad (LSN) y bilirrubina directa sérica ≤ 2 mg/dL. Se permitió el factor estimulante de colonias de granulocitos para los pacientes que desarrollaron neutropenia o fiebre en asociación con neutropenia. Las características basales de los pacientes y de la enfermedad se resumen en la Tabla 18.
Tabla 18: Características demográficas y relacionadas con la enfermedad al inicio del estudio en el estudio de MDS
a Categoría de riesgo IPSS: Baja (puntuación combinada = 0), Intermedia-1 (puntuación combinada = 0.5 a 1), Intermedia-2 (puntuación combinada = 1.5 a 2.0), Alta (puntuación combinada ≥ 2.5); Puntuación combinada = (Puntuación de blastocitos en médula ósea + Puntuación de cariotipo + Puntuación de citopenia). b Clasificación franco-estadounidense-británica (FAB) de MDS.
General
(N=148)
Edad (años)
Mediana
71
Mín., Máx.
37, 95
Sexo
n
(%)
Masculino
51
(34.5)
Femenino
97
(65.5)
Raza
n
(%)
Blanca
143
(96.6)
Otra
5
( 3.4)
Duración de los MDS (años)
Mediana
2.5
Mín., Máx.
0.1, 20.7
Anomalía citogenética Del 5 (q31-33)
n
(%)
Sí
148
(100)
Otras anomalías citogenéticas
37
(25.2)
Puntuación IPSS a
n
(%)
Baja (0)
55
(37.2)
Intermedia-1 (0.5-1.0)
65
(43.9)
Intermediate-2 (1.5-2.0)
6
( 4.1)
High (≥2.5)
2
( 1.4)
Missing
20
(13.5)
FAB Classification b from central review
n
(%)
RA
77
(52)
RARS
16
(10.8)
RAEB
30
(20.3)
CMML
3
( 2)
La frecuencia de independencia de transfusión de GR se evaluó utilizando criterios modificados de los criterios de respuesta del Grupo de Trabajo Internacional (IWG) para los SMD. La independencia de transfusión de GR se definió como la ausencia de cualquier transfusión de GR durante cualquier “período continuo” consecutivo de 56 días (8 semanas) durante el período de tratamiento.
Se observó independencia de transfusión en 99/148 (67 %) pacientes (IC del 95 % [59, 74]). La mediana de duración desde la fecha en que se declaró por primera vez la independencia de transfusión de GR (es decir, el último día del período sin transfusión de GR de 56 días) hasta la fecha en que se recibió una transfusión adicional después del período sin transfusión de 56 días entre los 99 respondedores fue de 44 semanas (rango de 0 a >67 semanas). El noventa por ciento de los pacientes que lograron un beneficio de transfusión lo hicieron al completar tres meses en el estudio.
Las tasas de independencia de transfusión de GR no se vieron afectadas por la edad ni el sexo.
La dosis de REVLIMID se redujo o interrumpió al menos una vez debido a un evento adverso en 118 (79,7 %) de los 148 pacientes; la mediana de tiempo hasta la primera reducción o interrupción de la dosis fue de 21 días (media, 35,1 días; rango, 2-253 días), y la mediana de duración de la primera interrupción de la dosis fue de 22 días (media, 28,5 días; rango, 2-265 días). Se requirió una segunda reducción o interrupción de la dosis debido a eventos adversos en 50 (33,8 %) de los 148 pacientes. El intervalo medio entre la primera y la segunda reducción o interrupción de la dosis fue de 51 días (media, 59,7 días; rango, 15-205 días) y la mediana de duración de la segunda interrupción de la dosis fue de 21 días (media, 26 días; rango, 2-148 días).
14.3 Linfoma de células del manto
Se realizó un ensayo multicéntrico, de un solo brazo y abierto de REVLIMID como agente único para evaluar la seguridad y eficacia de REVLIMID en pacientes con linfoma de células del manto que habían recaído después o eran refractarios a bortezomib o a un régimen que contenía bortezomib. Los pacientes con una depuración de creatinina ≥60 ml/min recibieron REVLIMID a una dosis de 25 mg una vez al día durante 21 días cada 28 días. Los pacientes con una depuración de creatinina ≥30 ml/min y <60 ml/min recibieron REVLIMID a una dosis de 10 mg una vez al día durante 21 días cada 28 días. El tratamiento se continuó hasta la progresión de la enfermedad, la toxicidad inaceptable o la retirada del consentimiento.
El ensayo incluyó a pacientes de al menos 18 años de edad con MCL confirmado por biopsia con enfermedad medible mediante tomografía computarizada. Los pacientes debían haber recibido tratamiento previo con una antraciclina o mitoxantrona, ciclofosfamida, rituximab y bortezomib, solos o en combinación. Los pacientes debían tener enfermedad refractaria documentada (definida como sin ninguna respuesta de RP o mejor durante el tratamiento con bortezomib o un régimen que contenía bortezomib), o enfermedad recidivante (definida como progresión dentro de un año después del tratamiento con bortezomib o un régimen que contenía bortezomib). Al momento del reclutamiento, los pacientes debían tener un recuento absoluto de neutrófilos (RAN) ≥1500/mm3, recuentos de plaquetas ≥60 000/mm3, SGOT/AST o SGPT/ALT séricas ≤3 x límite superior normal (LSN) a menos que hubiera evidencia documentada de afectación hepática por linfoma, bilirrubina total sérica ≤1,5 x LSN excepto en casos de síndrome de Gilbert o afectación hepática documentada por linfoma, y depuración de creatinina calculada (fórmula de Cockcroft-Gault) ≥30 ml/min.
La mediana de edad fue de 67 años (43-83), el 81 % eran hombres y el 96 % eran caucásicos. La siguiente tabla resume las características basales relacionadas con la enfermedad y el tratamiento anti-linfoma previo en el ensayo de linfoma de células del manto.
Tabla 19: Características basales relacionadas con la enfermedad y tratamiento anti-linfoma previo en el ensayo de linfoma de células del manto
a ECOG = Eastern Cooperative Oncology Group (Grupo Oncológico Cooperativo del Este). b MIPI = MCL International Prognostic Index (Índice Pronóstico Internacional del MCL). c La alta carga tumoral se define como al menos una lesión que tiene ≥5 cm de diámetro o 3 lesiones que tienen ≥3 cm de diámetro. d La enfermedad voluminosa se define como al menos una lesión que tiene ≥7 cm en el diámetro más largo.
Características basales de la enfermedad y tratamiento anti-linfoma previo
Total de pacientes
(N=134)
Estado de desempeño ECOGa n (%)
0
43 (32)
1
73 (54)
2
17 (13)
3
1 (<1)
Estadio avanzado de MCL, n (%)
III
27 (20)
IV
97 (72)
Puntuación MIPI alta o intermedia b, n (%)
90 (67)
Alta carga tumoralc, n (%)
77 (57)
Enfermedad voluminosad, n (%)
44 (33)
Enfermedad extranodal, n (%)
101 (75)
Número de terapias anti-linfoma sistémicas previas, n (%)
Mediana (rango)
4 (2, 10)
1
0 (0)
2
29 (22)
3
34 (25)
≥ 4
71 (53)
Número de sujetos que recibieron un régimen previo que contiene, n (%):
Anthracycline/mitoxantrone
133 (99)
Cyclophosphamide
133 (99)
Rituximab
134 (100)
Bortezomib
134 (100)
Refractario al Bortezomib previo, n (%)
81 (60)
Refractario a la última terapia previa, n (%)
74 (55)
Trasplante autólogo previo de médula ósea o células madre, n (%)
39 (29)
Los criterios de valoración de eficacia en el ensayo de MCL fueron la tasa de respuesta global (ORR) y la duración de la respuesta (DOR). La respuesta se determinó con base en la revisión de las exploraciones radiográficas por parte de un comité de revisión independiente de acuerdo con una versión modificada de los Criterios Internacionales de Respuesta al Linfoma en Talleres (Cheson, 1999). La DOR se define como el tiempo desde la respuesta inicial (al menos PR) hasta la progresión documentada de la enfermedad. Los resultados de eficacia para la población con MCL se basaron en todos los pacientes evaluables que recibieron al menos una dosis del fármaco del estudio y se presentan en la Tabla 20. El tiempo medio de respuesta fue de 2,2 meses (rango de 1,8 a 13 meses).
Tabla 20: Resultados de la respuesta en el ensayo fundamental del linfoma de células del manto
Análisis de respuesta (N = 133)
N (%)
95% CI
Tasa de respuesta global (IWRC) (CR + CRu +PR)
34 (26)
(18.4, 33.9)
Respuesta completa (CR + CRu)
9 (7)
(3.1, 12.5)
CR
1 (1)
CRu
8 (6)
Respuesta parcial (PR)
25 (19)
Duración de la respuesta (meses)
Mediana
95% CI
Duración de la respuesta global (CR + CRu + PR)
(N = 34)
16.6
(7.7, 26.7)
14.4 Linfoma folicular y de zona marginal
La eficacia de REVLIMID con rituximab en pacientes con linfoma folicular y de zona marginal en recaída o refractario se evaluó en los ensayos AUGMENT (NCT01938001) y MAGNIFY (NCT01996865).
AUGMENT es un ensayo aleatorizado, doble ciego y multicéntrico (n=358) en el que pacientes con linfoma folicular o de zona marginal en recaída o refractario fueron aleatorizados 1:1 para recibir REVLIMID y rituximab o rituximab y placebo. AUGMENT incluyó a pacientes diagnosticados con linfoma folicular de grado 1, 2 o 3a, que habían recibido al menos 1 tratamiento sistémico previo, eran refractarios o habían recaído, no eran refractarios a rituximab, tenían al menos una lesión ganglionar o extraganglionar medible mediante tomografía computarizada o resonancia magnética, y tenían una función adecuada de la médula ósea, el hígado y los riñones. La aleatorización se estratificó según el linfoma folicular frente al de zona marginal, el tratamiento previo con rituximab y el tiempo transcurrido desde otro tratamiento contra el linfoma. En AUGMENT, REVLIMID se administró por vía oral a una dosis de 20 mg una vez al día durante los días 1 a 21 de ciclos repetidos de 28 días durante un máximo de 12 ciclos o hasta que se produjera una toxicidad inaceptable. La dosis de rituximab fue de 375 mg/m2 cada semana en el ciclo 1 (días 1, 8, 15 y 22) y en el día 1 de cada ciclo de 28 días desde los ciclos 2 hasta el 5. Todos los cálculos de la dosis de rituximab se basaron en la superficie corporal (SC) del paciente, utilizando el peso real del paciente. Se permitieron ajustes de la dosis de REVLIMID en función de los hallazgos clínicos y de laboratorio. Un paciente con insuficiencia renal moderada (≥30 a <60 ml/minuto) recibió una dosis inicial más baja de REVLIMID de 10 mg diarios con la misma pauta. Después de 2 ciclos, la dosis de REVLIMID podía aumentarse a 15 mg una vez al día en los días 1 a 21 de cada ciclo de 28 días si el paciente toleraba la medicación.
MAGNIFY es un ensayo abierto y multicéntrico (n=232) en el que pacientes con linfoma folicular, de zona marginal o de células del manto en recaída o refractario recibieron 12 ciclos de inducción de REVLIMID y rituximab. MAGNIFY incluyó a pacientes diagnosticados con linfoma folicular de grado 1, 2, 3a, 3b (incluido el transformado), de zona marginal o de células del manto en estadio I a IV que habían sido tratados previamente para su linfoma, habían sido refractarios o habían recaído después de su último tratamiento, tenían al menos una lesión ganglionar o extraganglionar medible mediante tomografía computarizada o resonancia magnética, y tenían una función adecuada de la médula ósea, el hígado y los riñones. También se incluyeron pacientes refractarios a rituximab. La información de los sujetos que recibieron al menos 1 dosis de tratamiento inicial en los primeros 12 ciclos de inducción (n=222) en el ensayo MAGNIFY se incluyó en la evaluación de la eficacia de REVLIMID/rituximab en pacientes con linfoma folicular y de zona marginal en recaída o refractario. En MAGNIFY, se administraron 20 mg de REVLIMID en los días 1-21 de ciclos repetidos de 28 días durante un máximo de 12 ciclos o hasta que se produjera toxicidad inaceptable, progresión o retirada del consentimiento. La dosis de rituximab fue de 375 mg/m2 cada semana en el ciclo 1 (días 1, 8, 15 y 22) y en el día 1 de cada dos ciclos de 28 días (ciclos 3, 5, 7, 9 y 11) hasta 12 ciclos de tratamiento. Todos los cálculos de la dosis de rituximab se basaron en la SC del paciente y en el peso real. Se permitieron ajustes de la dosis en función de los hallazgos clínicos y de laboratorio.
Las características basales demográficas y relacionadas con la enfermedad en los ensayos AUGMENT y MAGNIFY se muestran en la siguiente tabla.
Tabla 21: Características demográficas y relacionadas con la enfermedad al inicio del estudio en pacientes con LF y LZM en los ensayos AUGMENT y MAGNIFY
Fecha de corte de datos: 22 de junio de 2018 (AUGMENT) y 1 de mayo de 2017 (MAGNIFY). a Definido por los criterios del GELF. b El paciente había recibido 0 (n=2) o 1 tratamiento sistémico previo. ECOG = Eastern Cooperative Oncology Group; FLIPI = índice pronóstico internacional del linfoma folicular
Parámetro
Ensayo AUGMENT
Ensayo MAGNIFY
REVLIMID + Rituximab
(N=178)
Rituximab + placebo
(grupo de control)
(N=180)
REVLIMID + Rituximab
(N=222)
Edad (años)
Mediana (máx., mín.)
64 (26, 86)
62 (35, 88)
65 (35, 91)
Distribución por edad, n (%)
<65 años
96 (54)
107 (59)
103 (46)
≥65 años
82 (46)
73 (41)
119 (54)
Sexo, n (%)
Masculino
75 (42)
97 (54)
122 (55)
Mujer
103 (58)
83 (46)
100 (45)
Raza
Blanca
118 (66)
115 (64)
206 (93)
Otras razas
54 (30)
64 (36)
14 (6)
No recogido o reportado
6 (3)
1 (0.6)
2 (1)
Superficie Corporal (BSA, m2)
Mediana (Máx., Mín.)
1.8 (1.4, 3.1)
1.8 (1.3, 2.7)
2 (1.3, 2.6)
Tipo de Enfermedad FL o LZM
Linfoma folicular
147 (83)
148 (82)
177 (80)
Linfoma de zona marginal
31 (17)
32 (18)
45 (20)
Subtipo de LZM al momento del diagnóstico (investigador), n (%)
MALT
14 (45)
16 (50)
10 (22)
Nodal
8 (26)
10 (31)
25 (56)
Esplénico
9 (29)
6 (19)
10 (22)
Estadio FL al momento del diagnóstico (investigador), n (%)
FL Grado 1-2
125 (85)
123 (83)
149 (84)
FL Grado 3a
22 (15)
25 (17)
28 (16)
Puntaje FLIPI al inicio (calculado), n (%)
No recogido
Riesgo bajo (0,1)
52 (29)
67 (37)
Riesgo intermedio (2)
55 (31)
58 (32)
Riesgo alto (≥3)
69 (39)
54 (30)
Perdido
2 (1)
1 (0.6)
Puntaje ECOG al inicio, n (%)
0
116 (65)
128 (71)
102 (46)
1
60 (34)
50 (28)
113 (51)
2
2 (1)
2 (1)
7 (3)
Alta carga tumorala al inicio del estudio, n (%)
Sí
97 (54)
86 (48)
148 (67)
No
81 (46)
94 (52)
74 (33)
Número de terapias sistémicas antilinfomatosas previas
1
102 (57)
97 (54)
94 (42)b
>1
76 (43)
83 (46)
128 (58)
En el estudio AUGMENT, se estableció la eficacia en la población por intención de tratar (ITT) con base en la supervivencia sin progresión determinada por un Comité de Revisión Independiente utilizando los criterios de respuesta modificados del Grupo de Trabajo Internacional de 2007. Los resultados de eficacia se resumen en la Tabla 22.
Tabla 22: Resultados de eficacia para pacientes en el ensayo AUGMENT (población con FL y MZL por ITT)
a La estimación de la mediana proviene del análisis de Kaplan-Meier. b El hazard ratio y su IC se estimaron a partir del modelo de riesgos proporcionales de Cox ajustado para la estratificación 3: tratamiento previo con rituximab (sí, no), tiempo transcurrido desde la última terapia antilinfoma (≤ 2, > 2 años) e histología de la enfermedad (FL, MZL). c Valor p de la prueba de log-rank estratificada por 3 factores mencionados anteriormente: tratamiento previo con rituximab (sí, no), tiempo transcurrido desde la última terapia antilinfoma (≤ 2, > 2 años) e histología de la enfermedad (FL, MZL). d Intervalo de confianza exacto para la distribución binomial.
Parámetro
REVLIMID + Rituximab
(N=178)
Rituximab + Placebo
(N=180)
PFS
Pacientes con evento, n (%)
68 (38.2)
115 (63.9)
Muerte
6 (8.8)
2 (1.7)
Progresión de la enfermedad
62 (91.2)
113 (98.3)
PFS, mediana a [95% CI] (meses)
39.4 [ 22.9, NE]
14.1 [11.4, 16.7]
HR b [95% CI]
0.46 [ 0.34, 0.62]
p-value c
<0.0001
Respuesta objetiva (CR+PR) , n(%) [95% CI] d
138 (77.5) [70.7, 83.4]
96 (53.3) [45.8, 60.8]
Curvas de Kaplan-Meier de supervivencia libre de progresión según la evaluación del IRC entre los brazos en el ensayo AUGMENT (población con LI y LZM ITT)
a = Los factores de estratificación incluyeron: tratamiento previo con rituximab (sí/no), tiempo desde la última terapia antilinfoma (≤2 años, >2 años) e histología de la enfermedad (FL o LZM). IC = intervalo de confianza; HR = razón de riesgo; KM = Kaplan-Meier; SLP = supervivencia libre de progresión
Linfoma folicular
En AUGMENT, la respuesta objetiva según la evaluación del IRC para pacientes con linfoma folicular fue del 80 % (118/147) [IC del 95 %: 73 %, 86 %]) en el brazo de REVLIMID con rituximab en comparación con el 55 % (82/148) [IC del 95 %: 47, 64] en el brazo de control.
En MAGNIFY, la respuesta global según la evaluación del investigador fue del 59 % (104/177) [IC del 95 %: 51, 66] para pacientes con linfoma folicular. No se alcanzó la mediana de duración de la respuesta con un tiempo medio de seguimiento de 7,9 meses [IC del 95 %: 4,6, 9,2].
Linfoma de la zona marginal
En AUGMENT, la respuesta objetiva según la evaluación del IRC para pacientes con linfoma de la zona marginal fue del 65 % (20/31) [IC del 95 %: 45 %, 81 %] en el brazo de REVLIMID con rituximab en comparación con el 44 % (14/32) [IC del 95 %: 26 %, 62 %] en el brazo de control.
En MAGNIFY, la respuesta global según la evaluación del investigador fue del 51 % (23/45) [IC del 95 %: 36, 66] para pacientes con linfoma de la zona marginal. No se alcanzó la mediana de duración de la respuesta con un tiempo medio de seguimiento de 11,5 meses [IC del 95 %: 8,0, 18,9].
15 REFERENCIAS
1. Drogas Peligrosas de OSHA. OSHA [Accedido el 29 de enero de 2013, desde http://www.osha.gov/SLTC/hazardousdrugs/index.html]
16 SUMINISTRO/ALMACENAMIENTO Y MANEJO
16.1 Forma de suministro
Cápsulas duras opacas blancas y azul verdosas impresas con “REV” en una mitad y “2.5 mg” en la otra mitad con tinta negra:
Frascos de 2.5 mg de 28
(NDC 59572-402-28)
Frascos de 2.5 mg de 100
(NDC 59572-402-00)
Cápsulas opacas blancas impresas con “REV” en una mitad y “5 mg” en la otra mitad con tinta negra:
Frascos de 5 mg de 28
(NDC 59572-405-28)
Frascos de 5 mg de 100
(NDC 59572-405-00)
Cápsulas opacas azul/verdes y amarillo pálido impresas con “REV” en una mitad y “10 mg” en la otra mitad con tinta negra:
Frascos de 10 mg de 28
(NDC 59572-410-28)
Frascos de 10 mg de 100
(NDC 59572-410-00)
Cápsulas opacas azul polvo y blancas impresas con “REV” en una mitad y “15 mg” en la otra mitad con tinta negra:
Frascos de 15 mg de 21
(NDC 59572-415-21)
Frascos de 15 mg de 100
(NDC 59572-415-00)
Cápsulas duras opacas azul polvo y azul verdosas impresas con “REV” en una mitad y “20 mg” en la otra mitad con tinta negra.
Frascos de 20 mg de 21
(NDC 59572-420-21)
Frascos de 20 mg de 100
(NDC 59572-420-00)
Cápsulas opacas blancas impresas con “REV” en una mitad y “25 mg” en la otra mitad con tinta negra:
Frascos de 25 mg de 21
(NDC 59572-425-21)
Frascos de 25 mg de 100
(NDC 59572-425-00)
16.2 Almacenamiento
Almacenar a 20°C – 25°C (68°F – 77°F); se permiten excursiones a 15°C – 30°C (59°F – 86°F) [Ver Temperatura ambiente controlada de USP].
16.3 Manejo y eliminación
Se debe tener cuidado al manipular REVLIMID. Las cápsulas de REVLIMID no deben abrirse ni romperse. Si el polvo de REVLIMID entra en contacto con la piel, lávese la piel inmediatamente y a fondo con agua y jabón. Si REVLIMID entra en contacto con las membranas mucosas, enjuague a fondo con agua.
Se deben considerar los procedimientos para el manejo y la eliminación adecuados de los medicamentos contra el cáncer. Se han publicado varias pautas sobre el tema.1
No dispense más de un suministro de 28 días.
17 INFORMACIÓN PARA EL PACIENTE
Aconseje al paciente que lea la información para pacientes aprobada por la FDA (Guía del medicamento)
Aconseje a las mujeres con potencial reproductivo que deben evitar el embarazo mientras toman REVLIMID y durante al menos 4 semanas después de completar el tratamiento.
•
Inicie el tratamiento con REVLIMID en mujeres con potencial reproductivo solo después de una prueba de embarazo negativa.
•
Aconseje a las mujeres con potencial reproductivo sobre la importancia de las pruebas de embarazo mensuales y la necesidad de utilizar 2 formas diferentes de anticoncepción, incluida al menos 1 forma altamente eficaz, simultáneamente durante el tratamiento con REVLIMID, durante la interrupción de la dosis y durante 4 semanas después de que haya terminado de tomar REVLIMID por completo. Las formas altamente eficaces de anticoncepción, aparte de la ligadura de trompas, incluyen el DIU y los métodos hormonales (píldoras anticonceptivas, inyecciones, parches o implantes) y la vasectomía de la pareja. Los métodos anticonceptivos eficaces adicionales incluyen el condón de látex o sintético, el diafragma y el capuchón cervical.
•
Indique a la paciente que deje de tomar REVLIMID inmediatamente y que se ponga en contacto con su proveedor de atención médica si se queda embarazada mientras toma este medicamento, si se retrasa su período menstrual o experimenta un sangrado menstrual inusual, si deja de tomar anticonceptivos o si cree, POR CUALQUIER MOTIVO, que puede estar embarazada.
Aconseje a los hombres que siempre usen un condón de látex o sintético durante cualquier contacto sexual con mujeres con potencial reproductivo mientras toman REVLIMID y hasta 4 semanas después de suspender REVLIMID, incluso si se han sometido a una vasectomía exitosa.
Se debe indicar a todos los pacientes que no donen sangre mientras toman REVLIMID, durante las interrupciones de la dosis y durante las 4 semanas siguientes a la suspensión de REVLIMID [ver Advertencias y precauciones (5.1)].
Programa REMS de lenalidomida
Debido al riesgo de toxicidad embrionaria y fetal, REVLIMID solo está disponible a través de un programa restringido llamado programa REMS de lenalidomida [ver Advertencias y precauciones (5.2)].
•
Los pacientes deben firmar un formulario de acuerdo entre el paciente y el médico y cumplir con los requisitos para recibir REVLIMID. En particular, las mujeres con potencial reproductivo deben cumplir con los requisitos de las pruebas de embarazo y la anticoncepción, y participar en encuestas telefónicas mensuales. Los hombres deben cumplir con los requisitos de anticoncepción [ver Uso en poblaciones específicas (8.3)].
•
REVLIMID solo está disponible en farmacias certificadas en el programa REMS de lenalidomida. Proporcione a los pacientes el número de teléfono y el sitio web para obtener información sobre cómo obtener el producto.
Registro de exposición durante el embarazo
Informe a las mujeres que existe un Registro de exposición durante el embarazo que monitorea los resultados del embarazo en mujeres expuestas a REVLIMID durante el embarazo y que pueden comunicarse con el Registro de exposición durante el embarazo llamando al 1-888-423-5436 [ver Uso en poblaciones específicas (8.1)].
Informe a los pacientes sobre el riesgo de trombosis, incluidos la TVP, la EP, el IM y el accidente cerebrovascular, y que informen inmediatamente cualquier signo y síntoma que sugiera estos eventos para su evaluación [ver Recuadro de advertencia y Advertencias y precauciones (5.4)].
Mayor mortalidad en pacientes con LLC
Informe a los pacientes que REVLIMID aumentó la mortalidad en pacientes con LLC y reacciones adversas cardiovasculares graves, como fibrilación auricular, infarto de miocardio e insuficiencia cardíaca [ver Advertencias y precauciones (5.5)].
Segundos tumores malignos primarios
Informe a los pacientes sobre el riesgo potencial de desarrollar segundos tumores malignos primarios durante el tratamiento con REVLIMID [ver Advertencias y precauciones (5.6)].
Hepatotoxicidad
Informe a los pacientes sobre el riesgo de hepatotoxicidad, incluida la insuficiencia hepática y la muerte, y que informen cualquier signo y síntoma asociado con este evento a su proveedor de atención médica para su evaluación [ver Advertencias y precauciones (5.8)].
Reacciones cutáneas graves
Informe a los pacientes sobre el riesgo potencial de reacciones cutáneas graves, como SJS, TEN y DRESS, e indíqueles que informen a su proveedor de atención médica sobre cualquier signo y síntoma asociado con estas reacciones para que los evalúe. Los pacientes con antecedentes de erupción cutánea de grado 4 asociada al tratamiento con talidomida no deben recibir REVLIMID [consulte Advertencias y precauciones (5.9)].
Síndrome de lisis tumoral
Informe a los pacientes sobre el riesgo potencial de síndrome de lisis tumoral y pídales que informen a su proveedor de atención médica sobre cualquier signo y síntoma asociado con este evento para que los evalúe [consulte Advertencias y precauciones (5.10)].
Reacción de exacerbación tumoral
Informe a los pacientes sobre el riesgo potencial de reacción de exacerbación tumoral y pídales que informen a su proveedor de atención médica sobre cualquier signo y síntoma asociado con este evento para que los evalúe [consulte Advertencias y precauciones (5.11)].
Informe a los pacientes sobre la posibilidad de reacciones de hipersensibilidad graves, como angioedema y anafilaxia, a REVLIMID. Indique a los pacientes que se comuniquen con su proveedor de atención médica de inmediato si presentan signos y síntomas de estas reacciones. Aconseje a los pacientes que busquen atención médica de emergencia si presentan signos o síntomas de reacciones de hipersensibilidad graves [consulte Advertencias y precauciones (5.15)].
REVLIMID debe tomarse una vez al día, aproximadamente a la misma hora todos los días,
•
REVLIMID puede tomarse con o sin alimentos.
•
Las cápsulas no deben abrirse, romperse ni masticarse. REVLIMID debe tragarse entero con agua.
•
Indique a los pacientes que, si olvidan una dosis de REVLIMID, pueden tomarla hasta 12 horas después de la hora habitual. Si han transcurrido más de 12 horas, se les debe indicar que se salten la dosis de ese día. Al día siguiente, deben tomar REVLIMID a la hora habitual. Advierta a los pacientes que no tomen 2 dosis para compensar la que olvidaron.
Comercializado por: Bristol-Myers Squibb Company Princeton, NJ 08543 USA
REVLIMID® es una marca comercial de Celgene Corporation, una empresa de Bristol-Myers Squibb.
RevPlyPI.031/MG.031
Guía de medicación
REVLIMID® (rev-li-mid)
(lenalidomida) cápsulas
¿Cuál es la información más importante que debo saber sobre REVLIMID?
Antes de comenzar a tomar REVLIMID, debe leer y aceptar todas las instrucciones del programa REMS de lenalidomida. Para obtener más información, llame al 1-888-423-5436 o visite www.lenalidomiderems.com. Antes de recetarle REVLIMID, su proveedor de atención médica le explicará el programa REMS de lenalidomida y le hará firmar el Formulario de Acuerdo Paciente-Médico. REVLIMID puede causar efectos secundarios graves que incluyen:
•
Posibles defectos de nacimiento (bebés deformados) o muerte de un bebé por nacer. Las mujeres embarazadas o que planean quedar embarazadas no deben tomar REVLIMID. REVLIMID es similar al medicamento talidomida. Sabemos que la talidomida puede causar defectos de nacimiento graves que ponen en peligro la vida. REVLIMID no se ha probado en mujeres embarazadas. REVLIMID ha dañado a animales no nacidos en pruebas con animales.
Las mujeres no deben quedar embarazadas:
o
Durante al menos 4 semanas antes de comenzar REVLIMID
o
Mientras toma REVLIMID
o
Durante cualquier interrupción (interrupciones) en su tratamiento con REVLIMID
o
Durante al menos 4 semanas después de dejar de tomar REVLIMID
Las mujeres que pueden quedar embarazadas:
o
Se realizarán pruebas de embarazo semanales durante 4 semanas, luego cada 4 semanas si su ciclo menstrual es regular, o cada 2 semanas si su ciclo menstrual es irregular.
o
Si pierde su período o tiene sangrado inusual, deberá hacerse una prueba de embarazo y recibir asesoramiento.
o
Debe aceptar usar dos formas aceptables de control de la natalidad al mismo tiempo, durante al menos 4 semanas antes, mientras toma, durante cualquier interrupción (interrupciones) en su tratamiento y durante al menos 4 semanas después de dejar de tomar REVLIMID.
o
Hable con su proveedor de atención médica para obtener información sobre las opciones de formas aceptables de control de la natalidad que puede usar para prevenir el embarazo antes, durante y después del tratamiento con REVLIMID.
o
Si tuvo relaciones sexuales sin protección o si cree que su método anticonceptivo ha fallado, deje de tomar REVLIMID inmediatamente y llame a su proveedor de atención médica de inmediato.
Si queda embarazada mientras toma REVLIMID, deje de tomarlo de inmediato y llame a su proveedor de atención médica. Si su proveedor de atención médica no está disponible, puede llamar al Centro de llamadas de REMS al 1-888-423-5436. Los proveedores de atención médica y los pacientes deben informar todos los casos de embarazo a:
o
FDA MedWatch al 1-800-FDA-1088, y
o
Centro de llamadas de REMS al 1-888-423-5436
Existe un registro de exposición al embarazo que monitorea los resultados de las mujeres que toman REVLIMID durante el embarazo, o si su pareja masculina toma REVLIMID y están expuestas durante el embarazo. Puede inscribirse en este registro llamando al Centro de llamadas de REMS al número de teléfono que se indica arriba.
REVLIMID puede pasar al semen humano:
o
Los hombres, incluidos los que se han sometido a una vasectomía, siempre deben usar un condón de látex o sintético durante cualquier contacto sexual con una mujer embarazada o una mujer que puede quedar embarazada mientras toma REVLIMID, durante cualquier interrupción (interrupciones) en su tratamiento con REVLIMID y hasta 4 semanas después de dejar de tomar REVLIMID.
o
No tenga relaciones sexuales sin protección con una mujer que esté o pueda quedar embarazada. Informe a su proveedor de atención médica si tiene relaciones sexuales sin protección con una mujer que esté o pueda quedar embarazada.
o
No done esperma mientras toma REVLIMID, durante cualquier interrupción (interrupciones) en su tratamiento y hasta 4 semanas después de dejar de tomar REVLIMID. Si una mujer queda embarazada con su esperma, el bebé puede estar expuesto a REVLIMID y puede nacer con defectos de nacimiento.
Hombres, si su pareja femenina queda embarazada, debe llamar a su proveedor de atención médica de inmediato.
•
Bajo recuento de glóbulos blancos (neutropenia) y bajo recuento de plaquetas (trombocitopenia). REVLIMID causa bajo recuento de glóbulos blancos y bajo recuento de plaquetas en la mayoría de las personas. Es posible que necesite una transfusión de sangre o ciertos medicamentos si su recuento sanguíneo baja demasiado. Su proveedor de atención médica debe controlar su recuento sanguíneo con frecuencia, especialmente durante los primeros meses de tratamiento con REVLIMID, y luego al menos mensualmente. Informe a su proveedor de atención médica si desarrolla algún sangrado o moretón durante el tratamiento con REVLIMID.
•
Coágulos de sangre. Los coágulos de sangre en las arterias, venas y pulmones ocurren con más frecuencia en las personas que toman REVLIMID. Este riesgo es aún mayor para las personas con mieloma múltiple que toman el medicamento dexametasona con REVLIMID. Los ataques cardíacos y los accidentes cerebrovasculares también ocurren con más frecuencia en las personas que toman REVLIMID con dexametasona. Para reducir este riesgo aumentado, la mayoría de las personas que toman REVLIMID también tomarán un medicamento anticoagulante. Antes de tomar REVLIMID, informe a su proveedor de atención médica:
o
Si ha tenido un coágulo de sangre en el pasado
o
Si tiene presión arterial alta, fuma o si le han dicho que tiene un nivel alto de grasa en la sangre (hiperlipidemia)
o
Sobre todos los medicamentos que toma. Ciertos otros medicamentos también pueden aumentar su riesgo de coágulos de sangre
Llame a su proveedor de atención médica o busque atención médica de inmediato si experimenta alguno de los siguientes durante el tratamiento con REVLIMID:
▪
Los signos o síntomas de un coágulo de sangre en el pulmón, brazo o pierna pueden incluir: dificultad para respirar, dolor en el pecho o hinchazón en el brazo o la pierna
▪
Los signos o síntomas de un ataque cardíaco pueden incluir: dolor en el pecho que puede extenderse a los brazos, cuello, mandíbula, espalda o área del estómago (abdomen), sensación de sudoración, dificultad para respirar, sensación de enfermedad o vómitos
▪
Los signos o síntomas de un derrame cerebral pueden incluir: entumecimiento o debilidad repentinos, especialmente en un lado del cuerpo, dolor de cabeza intenso o confusión, o problemas con la visión, el habla o el equilibrio
¿Qué es REVLIMID?
REVLIMID es un medicamento recetado, utilizado para tratar a adultos con:
•
mieloma múltiple (MM)
o
en combinación con el medicamento dexametasona, o
o
como tratamiento de mantenimiento después del trasplante autólogo de células madre hematopoyéticas (un tipo de trasplante de células madre que utiliza sus propias células madre)
•
una condición llamada síndromes mielodisplásicos (SMD). REVLIMID es para el tipo de SMD con un problema cromosómico donde falta parte del cromosoma 5. Este tipo de SMD se conoce como deleción 5q SMD. Las personas con este tipo de SMD pueden tener recuentos bajos de glóbulos rojos que requieren tratamiento con transfusiones de sangre.
•
linfoma de células del manto (LCM) cuando la enfermedad regresa o empeora después del tratamiento con 2 medicamentos previos, uno de los cuales incluía bortezomib. El LCM es un cáncer de un tipo de glóbulo blanco llamado linfocitos que se encuentran en los ganglios linfáticos.
•
linfoma folicular (LF) o linfoma de zona marginal (LMZ)
o
en combinación con un producto de rituximab, y
o
que han sido tratados previamente para su LF o LMZ
El LF y el LMZ son tipos de cáncer de glóbulos blancos llamados linfocitos B que se encuentran en los ganglios linfáticos y el bazo.
REVLIMID no debe utilizarse para tratar a personas que tienen leucemia linfocítica crónica (LLC) a menos que sean participantes en un ensayo clínico controlado. No se sabe si REVLIMID es seguro y eficaz en niños.
¿Quién no debe tomar REVLIMID?
No tome REVLIMID si usted:
•
está embarazada, planea quedar embarazada o queda embarazada durante el tratamiento con REVLIMID. Consulte “Cuál es la información más importante que debo saber sobre REVLIMID?”
•
es alérgico a la lenalidomida o a cualquiera de los ingredientes de REVLIMID. Consulte el final de esta Guía de medicamentos para obtener una lista completa de los ingredientes de REVLIMID.
¿Qué debo decirle a mi proveedor de atención médica antes de tomar REVLIMID?
Antes de tomar REVLIMID, informe a su proveedor de atención médica sobre todas sus afecciones médicas, incluso si usted:
•
tiene problemas hepáticos
•
tiene problemas renales o recibe tratamiento de diálisis renal
•
tiene problemas de tiroides
•
ha tenido una erupción cutánea grave con tratamiento con talidomida. No debe tomar REVLIMID.
•
es intolerante a la lactosa. REVLIMID contiene lactosa.
•
está amamantando. No amamante durante el tratamiento con REVLIMID. No se sabe si REVLIMID pasa a la leche materna y puede dañar a su bebé.
Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos herbales. REVLIMID y otros medicamentos pueden afectarse mutuamente, causando efectos secundarios graves. Hable con su proveedor de atención médica antes de tomar cualquier medicamento nuevo. Conozca los medicamentos que toma. Mantenga una lista de ellos para mostrársela a su proveedor de atención médica y farmacéutico.
¿Cómo debo tomar REVLIMID?
•
Tome REVLIMID exactamente como se lo recetaron y siga todas las instrucciones del programa Lenalidomide REMS
•
Trague las cápsulas de REVLIMID enteras con agua 1 vez al día. No abra, rompa ni mastique sus cápsulas.
•
REVLIMID se puede tomar con o sin alimentos.
•
Tome REVLIMID aproximadamente a la misma hora todos los días.
•
No abra ni rompa las cápsulas de REVLIMID ni las manipule más de lo necesario.
o
Si el polvo de la cápsula de REVLIMID entra en contacto con su piel, lávese la piel inmediatamente con agua y jabón.
o
Si el polvo de la cápsula de REVLIMID entra en contacto con el interior de sus ojos, nariz o boca, enjuague bien con agua.
•
Si olvida una dosis de REVLIMID y han pasado menos de 12 horas desde su hora habitual, tómela tan pronto como lo recuerde. Si han pasado más de 12 horas, simplemente omita la dosis olvidada. No tome 2 dosis al mismo tiempo.
•
Si toma demasiado REVLIMID, llame a su proveedor de atención médica de inmediato.
¿Qué debo evitar mientras tomo REVLIMID?
•
Consulte “Cuál es la información más importante que debo saber sobre REVLIMID?”
•
Mujeres: No quede embarazada y no amamante mientras toma REVLIMID.
•
Hombres: No done esperma mientras toma REVLIMID, durante cualquier interrupción (interrupciones) en su tratamiento y hasta 4 semanas después de dejar de tomar REVLIMID.
•
No comparta REVLIMID con otras personas. Puede causar defectos de nacimiento y otros problemas graves.
•
No done sangre mientras toma REVLIMID, durante cualquier interrupción (interrupciones) en su tratamiento y durante 4 semanas después de dejar de tomar REVLIMID. Si alguien que está embarazada recibe su sangre donada, su bebé puede estar expuesto a REVLIMID y puede nacer con defectos de nacimiento.
¿Cuáles son los posibles efectos secundarios de REVLIMID?
REVLIMID puede causar efectos secundarios graves, que incluyen:
•
Ver “Cuál es la información más importante que debo saber sobre REVLIMID?”
•
Aumento del riesgo de muerte en personas que tienen leucemia linfocítica crónica (LLC). Las personas con LLC que toman REVLIMID tienen un mayor riesgo de muerte en comparación con las personas que toman el medicamento clorambucil. REVLIMID puede causar problemas cardíacos graves que pueden provocar la muerte, incluida la fibrilación auricular, el ataque cardíaco o la insuficiencia cardíaca. No debe tomar REVLIMID si tiene LLC a menos que esté participando en un ensayo clínico controlado.
•
Riesgo de nuevos cánceres (malignidades). Se ha producido un aumento de nuevos cánceres (secundarios) en pacientes que recibieron REVLIMID y melfalán, o un trasplante de células madre de sangre, incluidos ciertos cánceres de sangre, como la leucemia mieloide aguda (LMA), y el síndrome mielodisplásico (SMD) y ciertos otros tipos de cánceres de la piel y otros órganos. Hable con su proveedor de atención médica sobre su riesgo de desarrollar nuevos cánceres si toma REVLIMID. Su proveedor de atención médica lo revisará para detectar nuevos cánceres durante su tratamiento con REVLIMID.
•
Problemas hepáticos graves, incluida la insuficiencia hepática y la muerte. Su proveedor de atención médica debe realizar análisis de sangre para verificar la función de su hígado durante su tratamiento con REVLIMID. Informe a su proveedor de atención médica de inmediato si desarrolla alguno de los siguientes síntomas de problemas hepáticos:
o
amarillento de su piel o la parte blanca de sus ojos (ictericia)
o
orina oscura o marrón (color té)
o
dolor en la parte superior derecha de su área estomacal (abdomen)
o
sangrado o moretones más fácilmente de lo normal
o
sentirse muy cansado
•
Reacciones cutáneas graves y reacciones alérgicas graves pueden ocurrir con REVLIMID y pueden causar la muerte.
Llame a su proveedor de atención médica de inmediato si desarrolla alguno de los siguientes signos o síntomas durante el tratamiento con REVLIMID:
o
una erupción cutánea roja y con picazón
o
descamación de su piel o ampollas
o
picazón severa
o
fiebre
Busque atención médica de emergencia de inmediato si desarrolla alguno de los siguientes signos o síntomas durante el tratamiento con REVLIMID:
o
hinchazón de sus labios, boca, lengua o garganta
o
dificultad para respirar o tragar
o
áreas rojas elevadas en su piel (urticaria)
o
un latido del corazón muy rápido
o
se siente mareado o débil
•
Síndrome de lisis tumoral (SLT). El SLT es causado por la rápida descomposición de las células cancerosas. El SLT puede causar insuficiencia renal y la necesidad de tratamiento de diálisis, ritmo cardíaco anormal, convulsiones y, a veces, la muerte. Su proveedor de atención médica puede realizar análisis de sangre para verificar si tiene SLT.
•
Empeoramiento de su tumor (reacción de brote tumoral) puede ocurrir con REVLIMID y puede causar la muerte. Informe a su proveedor de atención médica si tiene alguno de estos síntomas de reacción de brote tumoral durante el tratamiento con REVLIMID: ganglios linfáticos tiernos e inflamados, fiebre leve, dolor o erupción cutánea.
Su proveedor de atención médica puede decirle que disminuya su dosis, que deje de tomar REVLIMID temporalmente o que deje de tomarlo permanentemente si desarrolla ciertos efectos secundarios graves durante el tratamiento con REVLIMID.
•
Problemas de tiroides. Su proveedor de atención médica puede verificar la función de su tiroides antes de comenzar a tomar REVLIMID y durante el tratamiento con REVLIMID.
•
Riesgo de muerte temprana en MCL. En personas que tienen linfoma de células del manto (MCL), puede haber un riesgo de morir antes (muerte temprana) cuando toman REVLIMID. Hable con su proveedor de atención médica sobre cualquier inquietud y posibles factores de riesgo.
Los efectos secundarios más comunes de REVLIMID incluyen:
•
diarrea
•
erupción cutánea
•
náuseas
•
estreñimiento
•
cansancio o debilidad
•
fiebre
•
picazón
•
hinchazón de sus brazos, manos, piernas, pies y piel
•
problemas para dormir (insomnio)
•
dolor de cabeza
•
calambres o espasmos musculares
•
falta de aliento
•
tos, dolor de garganta y otros síntomas de resfriado
•
infección de las vías respiratorias superiores o bronquitis
•
inflamación del estómago y el intestino (“gripe estomacal”)
•
sangrado nasal
•
temblor o temblor (temblor)
•
dolores en las articulaciones
•
dolor en la espalda o en el área del estómago (abdomen)
Estos no son todos los posibles efectos secundarios de REVLIMID. Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088.
¿Cómo debo almacenar REVLIMID?
•
Almacene REVLIMID a temperatura ambiente entre 68 ° F y 77 ° F (20 ° C y 25 ° C).
•
Devuelva cualquier REVLIMID no utilizado a Bristol-Myers Squibb o a su proveedor de atención médica.
Mantenga REVLIMID y todos los medicamentos fuera del alcance de los niños.
Información general sobre el uso seguro y eficaz de REVLIMID.
Los medicamentos a veces se recetan para fines distintos de los que se enumeran en una Guía del medicamento. No tome REVLIMID para afecciones para las que no fue recetado. No le dé REVLIMID a otras personas, incluso si tienen los mismos síntomas que usted. Puede hacerles daño y puede causar defectos de nacimiento. Si desea obtener más información, hable con su proveedor de atención médica. Puede pedirle a su proveedor de atención médica o farmacéutico información sobre REVLIMID que esté escrita para profesionales de la salud.
Para obtener más información, llame al 1-888-423-5436 o visite www.lenalidomiderems.com.
¿Cuáles son los ingredientes de REVLIMID?
Ingrediente activo: lenalidomida Ingredientes inactivos: lactosa anhidra, celulosa microcristalina, croscarmelosa sódica y estearato de magnesio. La cápsula de 5 mg y 25 mg contiene gelatina, dióxido de titanio y tinta negra. La cápsula de 2.5 y 10 mg contiene gelatina, FD&C azul #2, óxido de hierro amarillo, dióxido de titanio y tinta negra. La cápsula de 15 mg contiene gelatina, FD&C azul #2, dióxido de titanio y tinta negra. La cápsula de 20 mg contiene gelatina, FD&C azul #2, óxido de hierro amarillo, dióxido de titanio y tinta negra. Comercializado por: Bristol-Myers Squibb Company, Princeton, NJ 08543 USA REVLIMID® es una marca registrada de Celgene Corporation, una compañía de Bristol-Myers Squibb. REVPlyMG.031 3/2023
Esta Guía de Medicamentos ha sido aprobada por la Administración de Alimentos y Medicamentos de los Estados Unidos. Revisado: MAR 2023
PANEL DE VISUALIZACIÓN PRINCIPAL DEL PAQUETE/ETIQUETA
NDC 59572-402-28
Revlimid
(lenalidomida) Cápsulas
2,5 mg
ADVERTENCIA: POTENCIAL PARA DEFECTOS CONGÉNITOS HUMANOS.
Sólo receta médica
28 Cápsulas
PANEL DE VISUALIZACIÓN PRINCIPAL DEL PAQUETE/ETIQUETA
NDC 59572-405-28
Revlimid
(lenalidomida) Cápsulas
5 mg
ADVERTENCIA: POSIBLE RIESGO DE DEFECTOS CONGÉNITOS.
Solo con receta médica
28 Cápsulas
PANEL DE VISUALIZACIÓN PRINCIPAL DEL PAQUETE/ETIQUETA
NDC 59572-410-28
Revlimid
(lenalidomide) Cápsulas
10 mg
ADVERTENCIA: POSIBILIDAD DE DEFECTOS DE NACIMIENTO HUMANOS.
Receta médica únicamente
28 Cápsulas
PANEL DE VISUALIZACIÓN PRINCIPAL DEL PAQUETE/ETIQUETA
NDC 59572-415-21
Revlimid
(lenalidomida) Cápsulas
15 mg
ADVERTENCIA: POSIBLE RIESGO DE DEFECTOS CONGÉNITOS.
Solo con receta médica
21 Cápsulas
PANEL DE VISUALIZACIÓN PRINCIPAL DEL PAQUETE/ETIQUETA
NDC 59572-420-21
Revlimid
(lenalidomida) Cápsulas
20 mg
ADVERTENCIA: POTENCIAL DE DEFECTOS CONGÉNITOS HUMANOS.
Sólo receta médica
21 Cápsulas
PANEL DE VISUALIZACIÓN PRINCIPAL DEL PAQUETE/ETIQUETA
NDC 59572-425-21
Revlimid
(lenalidomide) Cápsulas
25 mg
ADVERTENCIA: POSIBILIDAD DE DEFECTOS DE NACIMIENTO HUMANOS.
Fabricante de medicamentos: Boehringer Ingelheim Pharmaceuticals, Inc. (Updated: 2024-10-15) Tags: RANKL ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓNEstos aspectos destacados no incluyen toda la información necesaria para usar JARDIANCE de forma segura y eficaz. Consulte la información completa de prescripción de JARDIANCE. JARDIANCE® (comprimidos de empagliflozina), para administración…
Fabricante de medicamentos: GlaxoSmithKline Biologicals SA (Updated: 2023-05-22) Tags: 5-HT7 ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓNEstos puntos destacados no incluyen toda la información necesaria para usar SHINGRIX de manera segura y eficaz. Consulte la información de prescripción completa para SHINGRIX.SHINGRIX (vacuna recombinante contra el zóster, adyuvada), suspensión…
Fabricante de medicamentos: AstraZeneca Pharmaceuticals LP (Updated: 2024-09-17) Tags: FGF23Trop-2 ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓNEstos aspectos destacados no incluyen toda la información necesaria para usar FASENRA® de forma segura y eficaz. Consulte la información completa de prescripción de FASENRA.FASENRA (benralizumab) inyección, para uso subcutáneoAprobación inicial en…