Fabricante de medicamentos: Pharmacyclics LLC (Updated: 2024-12-20)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
IMBRUVICA® (ibrutinib) cápsulas, para uso oral
IMBRUVICA® (ibrutinib) tabletas, para uso oral
IMBRUVICA® (ibrutinib) suspensión oral
Aprobación inicial en EE. UU.: 2013
CAMBIOS RECIENTES IMPORTANTES
| Advertencias y precauciones, Hepatotoxicidad, Incluyendo Daño Hepático Inducido por Medicamentos (5.7) |
5/2024 |
INDICACIONES Y USO
IMBRUVICA es un inhibidor de la quinasa indicado para el tratamiento de:
- Pacientes adultos con leucemia linfocítica crónica (LLC)/linfoma linfocítico pequeño (LLP) (1.1).
- Pacientes adultos con leucemia linfocítica crónica (LLC)/linfoma linfocítico pequeño (LLP) con deleción 17p (1.2).
- Pacientes adultos con macroglobulinemia de Waldenström (MW) (1.3).
- Pacientes adultos y pediátricos de 1 año de edad y mayores con enfermedad crónica del injerto contra el huésped (EICHc) después del fracaso de una o más líneas de terapia sistémica (1.4).
DOSIFICACIÓN Y ADMINISTRACIÓN
- LLC/LLP y MW: 420 mg por vía oral una vez al día (2.1).
-
EICHc:
◦ Pacientes de 12 años y mayores: 420 mg por vía oral una vez al día (2.1).
◦ Pacientes de 1 a menos de 12 años de edad: 240 mg/m2 por vía oral una vez al día (hasta una dosis de 420 mg) (2.1).
Las tabletas o cápsulas deben tomarse por vía oral con un vaso de agua. No abra, rompa ni mastique las cápsulas. No corte, triture ni mastique las tabletas. Consulte la información de prescripción completa para obtener instrucciones de administración de la suspensión oral (2.1).
FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
CONTRAINDICACIONES
Ninguna (4)
ADVERTENCIAS Y PRECAUCIONES
- Hemorragia: Monitorear el sangrado y controlarlo (5.1).
- Infecciones: Monitorear a los pacientes para detectar fiebre e infecciones, evaluar con prontitud y tratar (5.2).
- Arritmias cardíacas, Insuficiencia cardíaca y Muerte súbita: Monitorear los síntomas de arritmias e insuficiencia cardíaca y controlarlos (5.3).
- Hipertensión: Monitorear la presión arterial y tratar (5.4).
- Citopenias: Revisar los hemogramas completos mensualmente (5.5).
- Segundas neoplasias malignas primarias: Se han producido otras neoplasias malignas en pacientes, incluidos cánceres de piel y otros carcinomas (5.6).
- Hepatotoxicidad, incluyendo daño hepático inducido por medicamentos: Monitorear la función hepática durante todo el tratamiento (5.7).
- Síndrome de lisis tumoral (SLT): Evaluar el riesgo inicial y tomar precauciones. Monitorear y tratar el SLT (5.8).
- Toxicidad embriofetal: Puede causar daño fetal. Aconsejar a las mujeres en edad fértil sobre el riesgo potencial para el feto y que usen métodos anticonceptivos eficaces (5.9, 8.1, 8.3).
REACCIONES ADVERSAS
- Las reacciones adversas más comunes (≥30%) en pacientes con neoplasias malignas de células B son trombocitopenia, diarrea, fatiga, dolor musculoesquelético, neutropenia, erupción cutánea, anemia, hematomas y náuseas (6).
- Las reacciones adversas más comunes (≥20%) en pacientes adultos o pediátricos con EICHc son fatiga, anemia, hematomas, diarrea, trombocitopenia, dolor musculoesquelético, pirexia, espasmos musculares, estomatitis, hemorragia, náuseas, dolor abdominal, neumonía y dolor de cabeza (6).
Para informar REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Pharmacyclics al 1-877-877-3536 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
INTERACCIONES CON OTROS MEDICAMENTOS
USO EN POBLACIONES ESPECÍFICAS
Consulte 17 para obtener INFORMACIÓN DE ASESORAMIENTO AL PACIENTE y el etiquetado del paciente aprobado por la FDA.
Revisado: 12/2024
Tabla de contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1
INDICACIONES Y USO
1.1
Leucemia Linfocítica Crónica/Linfoma Linfocítico Pequeño
1.2
Leucemia Linfocítica Crónica/Linfoma Linfocítico Pequeño con deleción 17p
1.3
Macroglobulinemia de Waldenström
1.4
Enfermedad crónica injerto contra huésped
2
POSOLOGÍA Y ADMINISTRACIÓN
2.1
Posología recomendada
2.2
Modificaciones de la dosis para reacciones adversas
2.3
Modificaciones de la dosis para uso con inhibidores de CYP3A
2.4
Modificaciones de la dosis para uso en insuficiencia hepática
3
FORMAS Y CONCENTRACIONES FARMACÉUTICAS
4
CONTRAINDICACIONES
5
ADVERTENCIAS Y PRECAUCIONES
5.1
Hemorragia
5.2
Infecciones
5.3
Arritmias cardíacas, insuficiencia cardíaca y muerte súbita
5.4
Hipertensión
5.5
Citopenias
5.6
Neoplasias malignas secundarias
5.7
Hepatotoxicidad, incluyendo lesión hepática inducida por fármacos
5.8
Síndrome de lisis tumoral
5.9
Toxicidad embriofetal
6
REACCIONES ADVERSAS
6.1
Experiencia en ensayos clínicos
6.2
Experiencia postcomercialización
7
INTERACCIONES MEDICAMENTOSAS
7.1
Efecto de los inhibidores de CYP3A sobre Ibrutinib
7.2
Efecto de los inductores de CYP3A sobre Ibrutinib
8
USO EN POBLACIONES ESPECÍFICAS
8.1
Embarazo
8.2
Lactancia
8.3
Mujeres y hombres en edad fértil
8.4
Uso pediátrico
8.5
Uso geriátrico
8.6
Insuficiencia hepática
8.7
Plasmaféresis
10
SOBREDOSIS
11
DESCRIPCIÓN
12
FARMACOLOGÍA CLÍNICA
12.1
Mecanismo de acción
12.2
Farmacodinamia
12.3
Farmacocinética
13
TOXICOLOGÍA NO CLÍNICA
13.1
Carcinogénesis, mutagénesis, deterioro de la fertilidad
14
ESTUDIOS CLÍNICOS
14.1
Leucemia Linfocítica Crónica/Linfoma Linfocítico Pequeño
14.2
Macroglobulinemia de Waldenström
14.3
Enfermedad crónica injerto contra huésped
16
PRESENTACIÓN/ALMACENAMIENTO Y MANEJO
17
INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no se enumeran.
1 INDICACIONES Y USO
1.1
Leucemia Linfocítica Crónica/Linfoma Linfocítico Pequeño
IMBRUVICA está indicado para el tratamiento de pacientes adultos con leucemia linfocítica crónica (LLC)/linfoma linfocítico pequeño (LLP).
1.2
Leucemia Linfocítica Crónica/Linfoma Linfocítico Pequeño con deleción 17p
IMBRUVICA está indicado para el tratamiento de pacientes adultos con leucemia linfocítica crónica (LLC)/linfoma linfocítico pequeño (LLP) con deleción 17p.
1.3
Macroglobulinemia de Waldenström
IMBRUVICA está indicado para el tratamiento de pacientes adultos con macroglobulinemia de Waldenström (MW).
1.4
Enfermedad crónica del injerto contra el huésped
IMBRUVICA está indicado para el tratamiento de pacientes adultos y pediátricos de 1 año o más con enfermedad crónica del injerto contra el huésped (EcICH) después del fracaso de una o más líneas de terapia sistémica.
2 DOSIS Y ADMINISTRACIÓN
2.1
Dosis Recomendada
Leucemia Linfocítica Crónica/Linfoma Linfocítico Pequeño y Macroglobulinemia de Waldenström
La dosis recomendada de IMBRUVICA para CLL/SLL y WM es de 420 mg por vía oral una vez al día hasta la progresión de la enfermedad o toxicidad inaceptable.
Para CLL/SLL, IMBRUVICA se puede administrar como agente único, en combinación con rituximab u obinutuzumab, o en combinación con bendamustina y rituximab (BR).
Para WM, IMBRUVICA se puede administrar como agente único o en combinación con rituximab.
Cuando se administra IMBRUVICA en combinación con rituximab u obinutuzumab, considere administrar IMBRUVICA antes de rituximab u obinutuzumab cuando se administran el mismo día.
Enfermedad Crónica del Injerto contra el Huésped
La dosis recomendada de IMBRUVICA para pacientes de 12 años o mayores con cGVHD es de 420 mg por vía oral una vez al día, y para pacientes de 1 a menos de 12 años con cGVHD es de 240 mg/m2 por vía oral una vez al día (hasta una dosis de 420 mg), hasta la progresión de la cGVHD, la recurrencia de una neoplasia subyacente o toxicidad inaceptable. Cuando un paciente ya no requiere terapia para el tratamiento de la cGVHD, se debe suspender IMBRUVICA considerando la evaluación médica del paciente individual.
| Dosis recomendada para alcanzar 240 mg/m2 | ||
| Rango de BSA* (m2) | Dosis (mg) de Cápsulas/Tabletas de IMBRUVICA a Administrar | Volumen (mL) de Suspensión Oral de IMBRUVICA (70 mg/mL) a Administrar |
| > 0.3 to 0.4 | – | 1.2 mL |
| > 0.4 to 0.5 | – | 1.5 mL |
| > 0.5 to 0.6 | – | 1.9 mL |
| > 0.6 to 0.7 | – | 2.2 mL |
| > 0.7 to 0.8 | 210 mg | 2.6 mL |
| > 0.8 to 0.9 | 210 mg | 2.9 mL |
| > 0.9 to 1 | 210 mg | 3.3 mL |
| > 1 to 1.1 | 280 mg | 3.6 mL |
| > 1.1 to 1.2 | 280 mg | 4 mL |
| > 1.2 to 1.3 | 280 mg | 4.3 mL |
| > 1.3 to 1.4 | 350 mg | 4.6 mL |
| > 1.4 to 1.5 | 350 mg | 5 mL |
| > 1.5 to 1.6 | 350 mg | 5.3 mL |
| > 1.6 | 420 mg | 6 mL |
*BSA = body surface area.
Administración
Administre IMBRUVICA aproximadamente a la misma hora todos los días.
Trague las tabletas o cápsulas enteras con un vaso de agua. No abra, rompa ni mastique las cápsulas. No corte, triture ni mastique las tabletas.
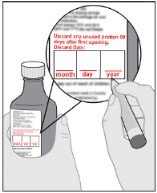
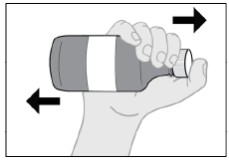
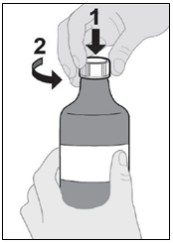
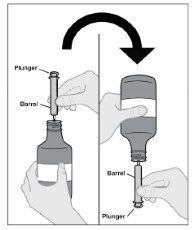
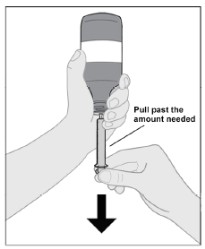
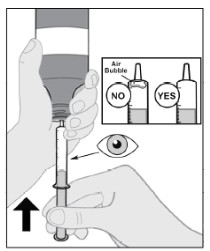
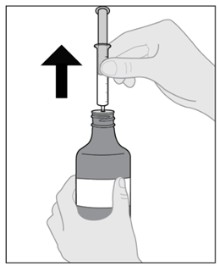
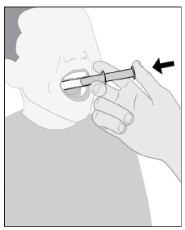
Siga las Instrucciones de Uso para obtener más detalles sobre la administración de la suspensión oral de IMBRUVICA.
Si no se toma una dosis de IMBRUVICA a la hora programada, se puede tomar lo antes posible el mismo día y volver al horario normal al día siguiente. No tome dosis adicionales de IMBRUVICA para compensar la dosis olvidada.
2.2
Modificaciones de la Dosis por Reacciones Adversas
Para las reacciones adversas enumeradas en la Tabla 2, interrumpa el tratamiento con IMBRUVICA. Una vez que la reacción adversa haya mejorado a Grado 1 o al valor inicial (recuperación), siga las modificaciones de dosis recomendadas (consulte la Tabla 2).
| Reacción Adversaa,b | Ocurrencia | Modificación de la Dosis para CLL/SLL, WM y Pacientes de 12 Años o Más con cGVHD Después de la Recuperación Dosis Inicial = 420 mg |
Modificación de la Dosis para Pacientes de 1 Año a Menos de 12 Años con cGVHD Después de la Recuperación Dosis Inicial = 240 mg/m2 |
| Insuficiencia cardíaca de Grado 2 | Primera | Reiniciar a 280 mg al díac | Reiniciar a 160 mg/m2 al díac |
| Segunda | Reiniciar a 140 mg al díac | Reiniciar a 80 mg/m2 al díac | |
| Tercera | Suspender IMBRUVICA | Suspender IMBRUVICA | |
| Arritmias cardíacas de Grado 3 | Primera | Reiniciar a 280 mg al díac | Reiniciar a 160 mg/m2 al díac |
| Segunda | Suspender IMBRUVICA | Suspender IMBRUVICA | |
| Insuficiencia cardíaca de Grado 3 o 4 Arritmias cardíacas de Grado 4 |
Primera | Suspender IMBRUVICA | Suspender IMBRUVICA |
| Otras toxicidades no hematológicas de Grado 3 o 4d Neutropenia de Grado 3 o 4 con infección o fiebre Toxicidades hematológicas de Grado 4 |
Primera | Reiniciar a 280 mg al día | Reiniciar a 160 mg/m2 al díac |
| Segunda | Reiniciar a 140 mg al día | Reiniciar a 80 mg/m2 al díac | |
| Tercera | Suspender IMBRUVICA | Suspender IMBRUVICA |
a [véase Advertencias y Precauciones (5)].
b Clasificación basada en los criterios del Instituto Nacional del Cáncer – Criterios de Terminología Común para Eventos Adversos (NCI-CTCAE) o en los criterios del Taller Internacional sobre Leucemia Linfocítica Crónica (iwCLL) para toxicidades hematológicas en LLC/SLL.
c Evaluar el beneficio-riesgo antes de reanudar el tratamiento.
d Para toxicidades no hematológicas de Grado 4, evaluar el beneficio-riesgo antes de reanudar el tratamiento.
| Dosis recomendada para alcanzar 160 mg/m2 | Dosis recomendada para alcanzar 80 mg/m2 | |||
| Rango de BSA* (m2) | Dosis (mg) de cápsulas/comprimidos de IMBRUVICA a administrar | Volumen (mL) de la suspensión oral de IMBRUVICA (70 mg/mL) a administrar | Dosis (mg) de cápsulas/comprimidos de IMBRUVICA a administrar | Volumen (mL) de la suspensión oral de IMBRUVICA (70 mg/mL) a administrar |
| > 0,3 a 0,4 | – | 0,8 mL | – | 0,4 mL |
| > 0,4 a 0,5 | – | 1 mL | – | 0,5 mL |
| > 0,5 a 0,6 | – | 1,3 mL | – | 0,6 mL |
| > 0,6 a 0,7 | – | 1,5 mL | – | 0,7 mL |
| > 0,7 a 0,8 | 140 mg | 1,7 mL | 70 mg | 0,9 mL |
| > 0,8 a 0,9 | 140 mg | 1,9 mL | 70 mg | 1 mL |
| > 0,9 a 1 | 140 mg | 2,2 mL | 70 mg | 1,1 mL |
| > 1 a 1,1 | 140 mg | 2,4 mL | 70 mg | 1,2 mL |
| > 1,1 a 1,2 | 210 mg | 2,6 mL | – | 1,3 mL |
| > 1,2 a 1,3 | 210 mg | 2,9 mL | – | 1,4 mL |
| > 1,3 a 1,4 | 210 mg | 3,1 mL | – | 1,5 mL |
| > 1,4 a 1,5 | 210 mg | 3,3 mL | 140 mg | 1,7 mL |
| > 1,5 a 1,6 | 280 mg | 3,5 mL | 140 mg | 1,8 mL |
| > 1,6 | 280 mg | 4 mL | 140 mg | 2 mL |
*BSA = área de superficie corporal.
2.3
Modificaciones de la dosis para el uso con inhibidores de CYP3A
Las modificaciones de dosis recomendadas se describen a continuación [ver Interacciones medicamentosas (7.1)]:
| Población de pacientes | Fármaco coadministrado | Dosis recomendada de IMBRUVICA |
| Neoplasias de células B |
|
280 mg una vez al día
Modifique la dosis según lo recomendado [ver Dosis y administración (2.2)]. |
|
140 mg una vez al día
Modifique la dosis según lo recomendado [ver Dosis y administración (2.2)]. |
|
|
70 mg una vez al día
Interrumpa la dosis según lo recomendado [ver Dosis y administración (2.2)]. |
|
|
Evitar el uso concomitante.
Si estos inhibidores se utilizarán a corto plazo (como antiinfecciosos durante siete días o menos), interrumpa IMBRUVICA. |
|
| Pacientes de 12 años o mayores con cGVHD |
|
420 mg una vez al día
Modifique la dosis según lo recomendado [ver Dosis y administración (2.2)]. |
|
280 mg una vez al día
Modifique la dosis según lo recomendado [ver Dosis y administración (2.2)]. |
|
|
140 mg una vez al día
Interrumpa la dosis según lo recomendado [ver Dosis y administración (2.2)]. |
|
|
Evitar el uso concomitante.
Si estos inhibidores se utilizarán a corto plazo (como antiinfecciosos durante siete días o menos), interrumpa IMBRUVICA. |
|
| Pacientes de 1 año a menos de 12 años de edad con cGVHD |
|
240 mg/m2 una vez al día
Modifique la dosis según lo recomendado [ver Dosis y administración (2.2)]. |
|
160 mg/m2 una vez al día | |
|
80 mg/m2 una vez al día | |
|
Evitar el uso concomitante.
Si estos inhibidores se utilizarán a corto plazo (como antiinfecciosos durante siete días o menos), interrumpa IMBRUVICA. |
Después de la suspensión de un inhibidor de CYP3A, reanude la dosis previa de IMBRUVICA [see Dosage and Administration (2.1), Drug Interactions (7.1)].
2.4
Modificaciones de la dosis para uso en insuficiencia hepática
Pacientes adultos con neoplasias malignas de células B
La dosis recomendada es de 140 mg al día para pacientes con insuficiencia hepática leve (clase A de Child-Pugh).
La dosis recomendada es de 70 mg al día para pacientes con insuficiencia hepática moderada (clase B de Child-Pugh).
Evite el uso de IMBRUVICA en pacientes con insuficiencia hepática grave (clase C de Child-Pugh) [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
Pacientes con cGVHD
La dosis recomendada es de 140 mg al día para pacientes de 12 años de edad y mayores con un nivel de bilirrubina total >1.5 a 3 x límite superior de lo normal (LSN) (a menos que sea de origen no hepático o debido al síndrome de Gilbert).
La dosis recomendada es de 80 mg/m2 al día para pacientes de 1 a menos de 12 años de edad con un nivel de bilirrubina total >1.5 a 3 x LSN (a menos que sea de origen no hepático o debido al síndrome de Gilbert).
Evite el uso de IMBRUVICA en estos pacientes con un nivel de bilirrubina total > 3 x LSN (a menos que sea de origen no hepático o debido al síndrome de Gilbert) [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
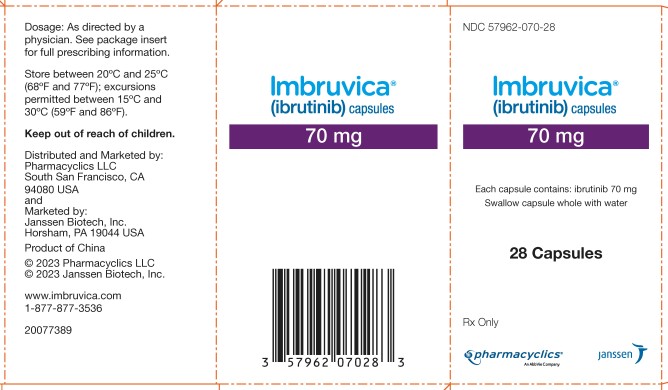
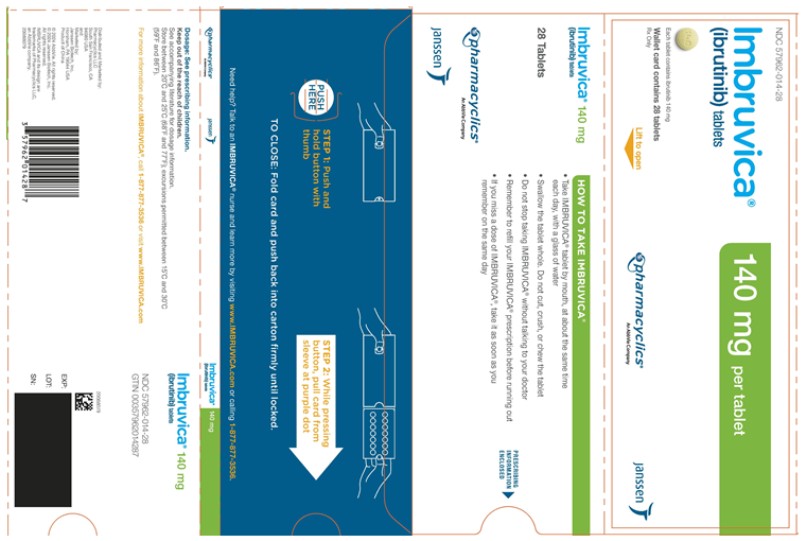
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Cápsulas:
Cada cápsula de 70 mg es una cápsula amarilla, opaca, marcada con “ibr 70 mg” en tinta negra.
Cada cápsula de 140 mg es una cápsula blanca, opaca, marcada con “ibr 140 mg” en tinta negra.
Comprimidos:
Cada comprimido de 140 mg es un comprimido redondo de color amarillo verdoso a verde, con “ibr” grabado en un lado y “140” en el otro.
Cada comprimido de 280 mg es un comprimido oblongo de color púrpura, con “ibr” grabado en un lado y “280” en el otro.
Cada comprimido de 420 mg es un comprimido oblongo de color amarillo verdoso a verde, con “ibr” grabado en un lado y “420” en el otro.
Suspensión oral:
70 mg/mL, suspensión blanca a blanquecina.
4 CONTRAINDICACIONES
Ninguna
5 ADVERTENCIAS Y PRECAUCIONES
5.1
Hemorragia
Se han producido eventos de hemorragia fatal en pacientes que recibieron IMBRUVICA. Se produjo hemorragia mayor (≥ Grado 3, grave o cualquier evento del sistema nervioso central; p. ej., hemorragia intracraneal [incluido hematoma subdural], hemorragia gastrointestinal, hematuria y hemorragia posprocedimiento) en el 4,2 % de los pacientes, con muertes en el 0,4 % de los 2838 pacientes que recibieron IMBRUVICA en 27 ensayos clínicos. Los eventos hemorrágicos de cualquier grado, incluidos hematomas y petequias, ocurrieron en el 39 %, y excluyendo hematomas y petequias ocurrieron en el 23 % de los pacientes que recibieron IMBRUVICA, respectivamente [véase Reacciones adversas (6.1)].
El mecanismo de los eventos hemorrágicos no se comprende bien.
El uso de agentes anticoagulantes o antiplaquetarios concomitantemente con IMBRUVICA aumenta el riesgo de hemorragia mayor. En los ensayos clínicos, el 3,1 % de los 2838 pacientes que recibieron IMBRUVICA sin terapia antiplaquetaria o anticoagulante experimentaron hemorragia mayor. La adición de terapia antiplaquetaria con o sin terapia anticoagulante aumentó este porcentaje al 4,4 %, y la adición de terapia anticoagulante con o sin terapia antiplaquetaria aumentó este porcentaje al 6,1 %. Considere los riesgos y beneficios de la terapia anticoagulante o antiplaquetaria cuando se coadministra con IMBRUVICA. Controle los signos y síntomas de hemorragia.
Considere la relación beneficio-riesgo de suspender IMBRUVICA durante al menos 3 a 7 días antes y después de la cirugía, según el tipo de cirugía y el riesgo de hemorragia [véase Estudios clínicos (14)].
5.2
Infecciones
Se han producido infecciones fatales y no fatales (incluidas las bacterianas, víricas o fúngicas) con el tratamiento con IMBRUVICA. Se produjeron infecciones de grado 3 o superior en el 21 % de los 1476 pacientes con neoplasias malignas de células B que recibieron IMBRUVICA en ensayos clínicos [véase Reacciones adversas (6.1, 6.2)]. Se han producido casos de leucoencefalopatía multifocal progresiva (LMP) y neumonía por Pneumocystis jirovecii (NJP) en pacientes tratados con IMBRUVICA. Considere la profilaxis según el estándar de atención en pacientes con mayor riesgo de infecciones oportunistas. Controle y evalúe a los pacientes en busca de fiebre e infecciones y trátelos adecuadamente.
5.3
Arritmias cardíacas, insuficiencia cardíaca y muerte súbita
Se han producido arritmias cardíacas y insuficiencia cardíaca fatales y graves con IMBRUVICA. Se produjeron muertes por causas cardíacas o muertes súbitas en el 1 % de los 4896 pacientes que recibieron IMBRUVICA en ensayos clínicos, incluso en pacientes que recibieron IMBRUVICA en monoterapia o regímenes de combinación no aprobados. Estas reacciones adversas se produjeron en pacientes con y sin hipertensión preexistente o comorbilidades cardíacas. Los pacientes con comorbilidades cardíacas pueden tener un mayor riesgo de estos eventos.
Se informaron taquiarritmias ventriculares de grado 3 o superior en el 0,2 %, fibrilación auricular y aleteo auricular de grado 3 o superior en el 3,7 %, e insuficiencia cardíaca de grado 3 o superior en el 1,3 % de los 4896 pacientes que recibieron IMBRUVICA en ensayos clínicos, incluso en pacientes que recibieron IMBRUVICA en monoterapia o regímenes de combinación no aprobados. Estos eventos se han producido particularmente en pacientes con factores de riesgo cardíaco, como hipertensión y diabetes mellitus, antecedentes de arritmias cardíacas y en pacientes con infecciones agudas [véase Reacciones adversas (6.1)].
Evalúe los antecedentes y la función cardíaca al inicio y controle a los pacientes en busca de arritmias cardíacas y función cardíaca. Obtenga una evaluación adicional (p. ej., ECG, ecocardiograma) según sea necesario para los pacientes que presenten síntomas de arritmia (p. ej., palpitaciones, mareos, síncope, dolor en el pecho), disnea de nueva aparición u otras preocupaciones cardiovasculares. Maneje las arritmias cardíacas y la insuficiencia cardíaca de manera adecuada, siga las pautas de modificación de la dosis [véase Posología y administración (2.2)] y considere los riesgos y beneficios del tratamiento continuado con IMBRUVICA.
5.4
Hipertensión
Se produjo hipertensión en el 19 % de los 1476 pacientes con neoplasias malignas de células B que recibieron IMBRUVICA en ensayos clínicos. La hipertensión de grado 3 o superior se produjo en el 8 % de los pacientes [véase Reacciones adversas (6.1)]. Según los datos de un subconjunto de estos pacientes (N = 1124), la mediana del tiempo hasta el inicio fue de 5,9 meses (rango, 0 a 24 meses). En un análisis de seguridad a largo plazo durante más de 5 años de 1284 pacientes con neoplasias malignas de células B tratados durante una mediana de 36 meses (rango, 0 a 98 meses), la tasa acumulativa de hipertensión aumentó con el tiempo. La prevalencia de hipertensión de grado 3 o superior fue del 4 % (año 0-1), 7 % (año 1-2), 9 % (año 2-3), 9 % (año 3-4) y 9 % (año 4-5); la incidencia general para el período de 5 años fue del 11 %.
Controle la presión arterial en los pacientes tratados con IMBRUVICA, inicie o ajuste la medicación antihipertensiva durante el tratamiento con IMBRUVICA según corresponda y siga las pautas de modificación de la dosis para la hipertensión de grado 3 o superior [véase Posología y administración (2.2)].
5.5
Citopenias
En 645 pacientes con neoplasias malignas de células B que recibieron IMBRUVICA como agente único, se produjo neutropenia de grado 3 o 4 en el 23% de los pacientes, trombocitopenia de grado 3 o 4 en el 8% y anemia de grado 3 o 4 en el 2,8%, según las mediciones de laboratorio [véase Reacciones adversas (6.1)].
Controlar los hemogramas completos mensualmente.
5.6
Neoplasias malignas secundarias
Se produjeron otras neoplasias malignas (10%), incluidos carcinomas no cutáneos (3,9%), entre los 1476 pacientes con neoplasias malignas de células B que recibieron IMBRUVICA en ensayos clínicos [véase Reacciones adversas (6.1)]. La neoplasia maligna secundaria más frecuente fue el cáncer de piel no melanoma (6%).
5.7
Hepatotoxicidad, incluida la lesión hepática inducida por fármacos
Se ha producido hepatotoxicidad, incluidos casos graves, potencialmente mortales y potencialmente mortales de lesión hepática inducida por fármacos (DILI), en pacientes tratados con inhibidores de la tirosina quinasa de Bruton, incluido IMBRUVICA.
Evaluar la bilirrubina y las transaminasas al inicio y durante todo el tratamiento con IMBRUVICA. En los pacientes que desarrollan pruebas hepáticas anormales después de IMBRUVICA, controlar con más frecuencia las anomalías de las pruebas hepáticas y los signos y síntomas clínicos de toxicidad hepática. Si se sospecha DILI, suspender IMBRUVICA. Tras la confirmación de DILI, interrumpir el tratamiento con IMBRUVICA.
5.8
Síndrome de lisis tumoral
Se ha notificado con poca frecuencia el síndrome de lisis tumoral con IMBRUVICA [véase Reacciones adversas (6.2)]. Evaluar el riesgo basal (p. ej., alta carga tumoral) y tomar las precauciones adecuadas. Controlar estrechamente a los pacientes y tratarlos según corresponda.
5.9
Toxicidad embriofetal
Según los hallazgos en animales, IMBRUVICA puede causar daño fetal cuando se administra a una mujer embarazada. La administración de ibrutinib a ratas y conejas embarazadas durante el período de organogénesis provocó toxicidad embriofetal, incluidas malformaciones, a exposiciones que fueron de 3 a 20 veces superiores a las notificadas en pacientes con neoplasias hematológicas. Advertir a las mujeres embarazadas del riesgo potencial para el feto. Aconsejar a las mujeres en edad fértil que utilicen un método anticonceptivo eficaz durante el tratamiento con IMBRUVICA y durante 1 mes después de la última dosis. [véase Uso en poblaciones específicas (8.1)].
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas clínicamente significativas se describen en otras partes del etiquetado:
- Hemorrhage [see Warnings and Precautions (5.1)]
- Infections [see Warnings and Precautions (5.2)]
- Cardiac Arrhythmias, Cardiac Failure, and Sudden Death [see Warnings and Precautions (5.3)]
- Hypertension [see Warnings and Precautions (5.4)]
- Cytopenias [see Warnings and Precautions (5.5)]
- Second Primary Malignancies [see Warnings and Precautions (5.6)]
- Hepatotoxicity, including DILI [see Warnings and Precautions (5.7)]
- Tumor Lysis Syndrome [see Warnings and Precautions (5.8)]
6.1
Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se realizan bajo condiciones ampliamente variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un medicamento no se pueden comparar directamente con las tasas de ensayos clínicos de otro medicamento y es posible que no reflejen las tasas observadas en la práctica.
A menos que se especifique lo contrario, la población de seguridad combinada descrita en las ADVERTENCIAS Y PRECAUCIONES refleja la exposición a IMBRUVICA en 6 ensayos. IMBRUVICA se administró como agente único a 420 mg por vía oral una vez al día (475 pacientes), como agente único a 560 mg por vía oral una vez al día [1,3 veces la dosis recomendada para adultos (174 pacientes)] y en combinación con otros medicamentos a 420 mg por vía oral una vez al día (827 pacientes) en pacientes con neoplasias malignas de células B. En esta población de seguridad combinada de 1476 pacientes, el 87 % estuvo expuesto durante 6 meses o más y el 68 % estuvo expuesto durante más de un año. Las reacciones adversas más comunes (≥ 30 %) fueron trombocitopenia, diarrea, fatiga, dolor musculoesquelético, neutropenia, erupción cutánea, anemia, hematomas y náuseas.
Ciertas subsecciones en las ADVERTENCIAS Y PRECAUCIONES incluyen pacientes que recibieron IMBRUVICA en regímenes de monoterapia o combinación no aprobados.
Leucemia linfocítica crónica/linfoma linfocítico pequeño
Los datos que se describen a continuación reflejan la exposición a IMBRUVICA en un ensayo clínico abierto de un solo brazo (Estudio 1102) y cinco ensayos clínicos controlados aleatorizados (RESONATE, RESONATE-2, HELIOS, iLLUMINATE y E1912) en pacientes con CLL/SLL (n=2016 en total, incluidos n=1133 pacientes expuestos a IMBRUVICA). En general, los pacientes con aclaramiento de creatinina (CLcr) ≤ 30 ml/min, AST o ALT ≥ 2,5 x LSN o bilirrubina total ≥ 1,5 x LSN (a menos que sea de origen no hepático) fueron excluidos de estos ensayos. En el estudio E1912, se excluyeron los pacientes con AST o ALT > 3 x LSN o bilirrubina total > 2,5 x LSN. El estudio 1102 incluyó a 51 pacientes con CLL/SLL previamente tratados. RESONATE incluyó a 386 pacientes aleatorizados con CLL o SLL previamente tratados que recibieron IMBRUVICA como agente único o ofatumumab. RESONATE-2 incluyó a 267 pacientes aleatorizados con CLL o SLL sin tratamiento previo que tenían 65 años o más y recibieron IMBRUVICA como agente único o clorambucilo. HELIOS incluyó a 574 pacientes aleatorizados con CLL o SLL previamente tratados que recibieron IMBRUVICA en combinación con BR o placebo en combinación con BR. iLLUMINATE incluyó a 228 pacientes aleatorizados con CLL/SLL sin tratamiento previo que tenían 65 años o más o con afecciones médicas coexistentes y recibieron IMBRUVICA en combinación con obinutuzumab o clorambucilo en combinación con obinutuzumab. E1912 incluyó a 510 pacientes con CLL/SLL no tratados previamente que tenían 70 años o menos y recibieron IMBRUVICA en combinación con rituximab o recibieron fludarabina, ciclofosfamida y rituximab (FCR).
Las reacciones adversas más comunes en pacientes con CLL/SLL que recibieron IMBRUVICA (≥ 30 %) fueron trombocitopenia, diarrea, fatiga, dolor musculoesquelético, neutropenia, erupción cutánea, anemia, hematomas y náuseas.
Entre el cuatro y el diez por ciento de los pacientes con CLL/SLL que recibieron IMBRUVICA interrumpieron el tratamiento debido a reacciones adversas. Estas incluyeron neumonía, hemorragia, fibrilación auricular, neutropenia, artralgia, erupción cutánea y trombocitopenia. Las reacciones adversas que llevaron a la reducción de la dosis ocurrieron en aproximadamente el 9 % de los pacientes.
Estudio 1102
Las reacciones adversas y las anomalías de laboratorio del Estudio 1102 (N=51) con IMBRUVICA 420 mg diarios como agente único en pacientes con CLL/SLL previamente tratados que ocurren a una tasa de ≥ 10 % con una mediana de duración del tratamiento de 15,6 meses se presentan en la Tabla 5 y la Tabla 6.
| Sistema corporal | Reacción adversa | Todos los grados (%) | Grado 3 o superior (%) |
| Trastornos gastrointestinales | Diarrea Estreñimiento Náuseas Estomatitis Vómitos Dolor abdominal Dispepsia |
59 22 20 20 18 14 12 |
4 2 2 0 2 0 0 |
| Trastornos de la piel y del tejido subcutáneo | Moretones Erupción cutánea Petequias |
51 25 16 |
2 0 0 |
| Infecciones e infestaciones | Infección de las vías respiratorias superiores Sinusitis Infección de la piel Neumonía Infección del tracto urinario |
47 22 16 12 12 |
2 6 6 10 2 |
| Trastornos generales y afecciones en el lugar de administración | Fatiga Pirexia Edema periférico Astenia Escalofríos |
33 24 22 14 12 |
6 2 0 6 0 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | Dolor musculoesquelético Artralgia Espasmos musculares |
25 24 18 |
6 0 2 |
| Trastornos respiratorios, torácicos y mediastínicos | Tos Dolor orofaríngeo Disnea |
22 14 12 |
0 0 0 |
| Trastornos del sistema nervioso | Mareos Dolor de cabeza |
20 18 |
0 2 |
| Trastornos vasculares | Hipertensión | 16 | 8 |
| Trastornos del metabolismo y la nutrición | Disminución del apetito | 16 | 2 |
| Neoplasias benignas, malignas, no especificadas | Segundas neoplasias malignas | 10 | 2† |
†Una muerte de paciente debido a sarcoma histiocítico.
| Porcentaje de pacientes (N=51) | ||
| Todos los grados (%) | Grado 3 o 4 (%) | |
| Disminución de plaquetas | 69 | 12 |
| Disminución de neutrófilos | 53 | 26 |
| Disminución de hemoglobina | 43 | 0 |
* Basado en mediciones de laboratorio según los criterios de IWCLL y las reacciones adversas.
Se produjo trombocitopenia de grado 4 emergente del tratamiento (8%) y neutropenia (12%) en los pacientes.
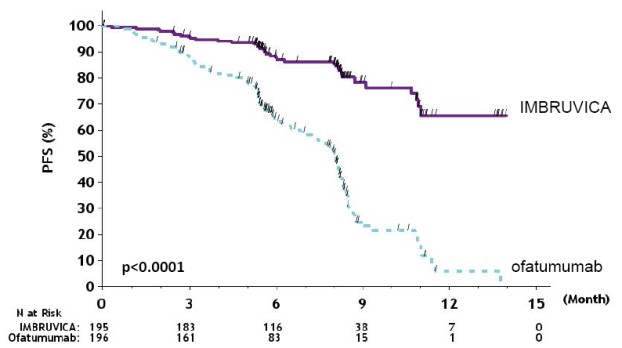
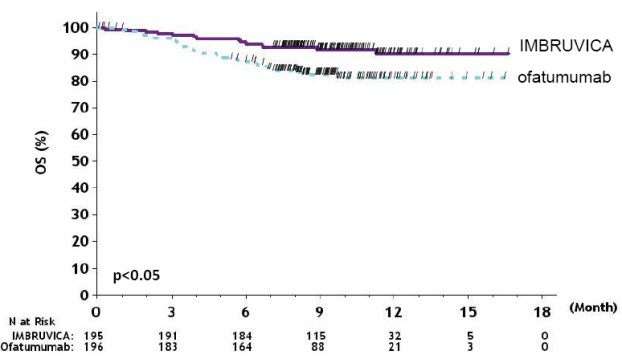
RESONATE
Las reacciones adversas y las anomalías de laboratorio que se describen a continuación en la Tabla 7 y la Tabla 8 reflejan la exposición a IMBRUVICA con una duración media de 8.6 meses y la exposición a ofatumumab con una mediana de 5.3 meses en RESONATE en pacientes con CLL/SLL previamente tratados.
| Sistema del cuerpo Reacción adversa |
IMBRUVICA (N=195) |
Ofatumumab (N=191) |
||
| Todos los grados (%) |
Grado 3 o Superior (%) |
Todos los grados (%) |
Grado 3 o Superior (%) |
|
| Trastornos gastrointestinales | ||||
| Diarrea | 48 | 4 | 18 | 2 |
| Náuseas | 26 | 2 | 18 | 0 |
| Estomatitis* | 17 | 1 | 6 | 1 |
| Estreñimiento | 15 | 0 | 9 | 0 |
| Vómitos | 14 | 0 | 6 | 1 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | ||||
| Dolor musculoesquelético* | 28 | 2 | 18 | 1 |
| Artralgia | 17 | 1 | 7 | 0 |
| Espasmos musculares | 13 | 0 | 8 | 0 |
| Trastornos de la piel y del tejido subcutáneo | ||||
| Erupción cutánea* | 24 | 3 | 13 | 0 |
| Petequias | 14 | 0 | 1 | 0 |
| Hematomas* | 12 | 0 | 1 | 0 |
| Trastornos generales y alteraciones en el lugar de administración | ||||
| Pirexia | 24 | 2 | 15 | 2† |
| Trastornos respiratorios, torácicos y mediastínicos | ||||
| Tos | 19 | 0 | 23 | 1 |
| Disnea | 12 | 2 | 10 | 1 |
| Infecciones e infestaciones | ||||
| Infección de las vías respiratorias superiores | 16 | 1 | 11 | 2† |
| Pneumonia* | 15 | 12† | 13 | 10† |
| Sinusitis* | 11 | 1 | 6 | 0 |
| Infección del tracto urinario | 10 | 4 | 5 | 1 |
| Trastornos del sistema nervioso | ||||
| Dolor de cabeza | 14 | 1 | 6 | 0 |
| Mareo | 11 | 0 | 5 | 0 |
| Lesiones, intoxicaciones y complicaciones de procedimientos | ||||
| Contusión | 11 | 0 | 3 | 0 |
| Trastornos oculares | ||||
| Visión borrosa | 10 | 0 | 3 | 0 |
| El sistema del cuerpo y los términos individuales de reacciones adversas a medicamentos (RAM) se ordenan en orden descendente de frecuencia en el brazo de IMBRUVICA. * Incluye múltiples términos de RAM. † Incluye 3 eventos de neumonía con desenlace fatal en cada brazo y 1 evento de pirexia e infección de las vías respiratorias superiores con desenlace fatal en el brazo de ofatumumab. |
||||
| IMBRUVICA (N=195) |
Ofatumumab (N=191) |
|||
| Todos los grados (%) |
Grado 3 o 4 (%) |
Todos los grados (%) |
Grado 3 o 4 (%) |
|
| Disminución de neutrófilos | 51 | 23 | 57 | 26 |
| Disminución de plaquetas | 52 | 5 | 45 | 10 |
| Disminución de hemoglobina | 36 | 0 | 21 | 0 |
Se presentó trombocitopenia de Grado 4 emergente del tratamiento (2% en el brazo de IMBRUVICA vs 3% en el brazo de ofatumumab) y neutropenia (8% en el brazo de IMBRUVICA vs 8% en el brazo de ofatumumab) en los pacientes.
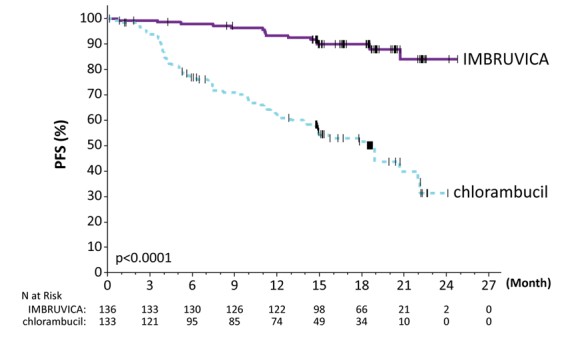
RESONATE-2
Las reacciones adversas y las anomalías de laboratorio que se describen a continuación en la Tabla 9 y la Tabla 10 reflejan la exposición a IMBRUVICA con una duración media de 17.4 meses. La mediana de exposición a clorambucilo fue de 7.1 meses en RESONATE-2.
| Sistema del cuerpo Reacción adversa |
IMBRUVICA (N=135) |
Clorambucilo (N=132) |
||
| Todos los grados (%) |
Grado 3 o superior (%) | Todos los grados (%) |
Grado 3 o superior (%) | |
| Trastornos gastrointestinales | ||||
| Diarrea | 42 | 4 | 17 | 0 |
| Náuseas | 22 | 1 | 39 | 1 |
| Estreñimiento | 16 | 1 | 16 | 0 |
| Estomatitis* | 14 | 1 | 4 | 1 |
| Vómitos | 13 | 0 | 20 | 1 |
| Dolor abdominal | 13 | 3 | 11 | 1 |
| Dispepsia | 11 | 0 | 2 | 0 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | ||||
| Dolor musculoesquelético* | 36 | 4 | 20 | 0 |
| Artralgia | 16 | 1 | 7 | 1 |
| Espasmos musculares | 11 | 0 | 5 | 0 |
| Trastornos generales y alteraciones en el lugar de administración | ||||
| Fatiga | 30 | 1 | 38 | 5 |
| Edema periférico | 19 | 1 | 9 | 0 |
| Pirexia | 17 | 0 | 14 | 2 |
| Trastornos respiratorios, torácicos y mediastínicos | ||||
| Tos | 22 | 0 | 15 | 0 |
| Disnea | 10 | 1 | 10 | 0 |
| Trastornos de la piel y del tejido subcutáneo | ||||
| Erupción cutánea* | 21 | 4 | 12 | 2 |
| Moretones* | 19 | 0 | 7 | 0 |
| Trastornos oculares | ||||
| Sequedad ocular | 17 | 0 | 5 | 0 |
| Aumento del lagrimeo | 13 | 0 | 6 | 0 |
| Visión borrosa | 13 | 0 | 8 | 0 |
| Agudeza visual reducida | 11 | 0 | 2 | 0 |
| Infecciones e infestaciones | ||||
| Infección de las vías respiratorias superiores | 17 | 2 | 17 | 2 |
| Infección de la piel* | 15 | 2 | 3 | 1 |
| Neumonía* | 14 | 8 | 7 | 4 |
| Infecciones del tracto urinario | 10 | 1 | 8 | 1 |
| Trastornos vasculares | ||||
| Hipertensión* | 14 | 4 | 1 | 0 |
| Trastornos del sistema nervioso | ||||
| Dolor de cabeza | 12 | 1 | 10 | 2 |
| Mareos | 11 | 0 | 12 | 1 |
| Investigaciones | ||||
| Pérdida de peso | 10 | 0 | 12 | 0 |
Los sujetos con múltiples eventos para un término de RA dado se cuentan solo una vez para cada término de RA.
El sistema del cuerpo y los términos individuales de RA se ordenan en orden descendente de frecuencia en el brazo de IMBRUVICA.
* Incluye múltiples términos de RA.
| IMBRUVICA (N=135) |
Chlorambucil (N=132) |
|||
| Todos los Grados (%) |
Grado 3 o 4 (%) |
Todos los Grados (%) |
Grado 3 o 4 (%) |
|
| Neutrophils Decreased | 55 | 28 | 67 | 31 |
| Platelets Decreased | 47 | 7 | 58 | 14 |
| Hemoglobin Decreased | 36 | 0 | 39 | 2 |
Se produjo trombocitopenia de Grado 4 emergente del tratamiento (1% en el brazo de IMBRUVICA vs 3% en el brazo de clorambucilo) y neutropenia (11% en el brazo de IMBRUVICA vs 12% en el brazo de clorambucilo) en pacientes.
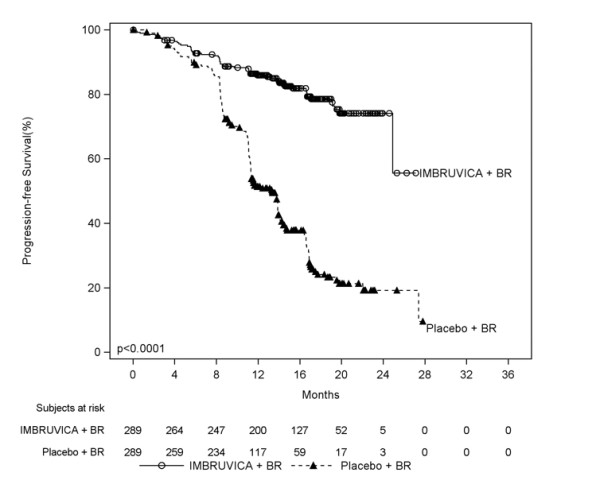
HELIOS
Las reacciones adversas descritas a continuación en la Tabla 11 reflejan la exposición a IMBRUVICA + BR con una duración media de 14,7 meses y la exposición a placebo + BR con una mediana de 12,8 meses en HELIOS en pacientes con CLL/SLL previamente tratados.
| Sistema Corporal Reacción Adversa |
IMBRUVICA + BR (N=287) |
Placebo + BR (N=287) |
||
| Todos los Grados (%) |
Grado 3 o Superior (%) | Todos los Grados (%) |
Grado 3 o Superior (%) | |
| Trastornos sanguíneos y del sistema linfático | ||||
| Neutropenia* | 66 | 61 | 60 | 56† |
| Trombocitopenia* | 34 | 16 | 26 | 16 |
| Trastornos gastrointestinales | ||||
| Diarrea | 36 | 2 | 23 | 1 |
| Dolor abdominal | 12 | 1 | 8 | <1 |
| Trastornos de la piel y del tejido subcutáneo | ||||
| Erupción cutánea* | 32 | 4 | 25 | 1 |
| Moretones * | 20 | <1 | 8 | <1 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | ||||
| Dolor musculoesquelético* | 29 | 2 | 20 | 0 |
| Espasmos musculares | 12 | <1 | 5 | 0 |
| Trastornos generales y afecciones en el lugar de administración | ||||
| Pirexia | 25 | 4 | 22 | 2 |
| Trastornos vasculares | ||||
| Hemorragia* | 19 | 2† | 9 | 1 |
| Hipertensión* | 11 | 5 | 5 | 2 |
| Infecciones e infestaciones | ||||
| Bronquitis | 13 | 2 | 10 | 3 |
| Infección de la piel* | 10 | 3 | 6 | 2 |
| Trastornos del metabolismo y la nutrición | ||||
| Hiperuricemia | 10 | 2 | 6 | 0 |
El sistema del cuerpo y los términos individuales de las reacciones adversas al medicamento (RAM) se clasifican en orden descendente de frecuencia en el grupo de IMBRUVICA.
* Incluye múltiples términos de RAM.
<1 se utiliza para una frecuencia superior a 0 e inferior al 0,5%.
† Incluye 2 eventos de hemorragia con resultado fatal en el grupo de IMBRUVICA y 1 evento de neutropenia con resultado fatal en el grupo de placebo + BR.
La fibrilación auricular de cualquier grado ocurrió en el 7% de los pacientes tratados con IMBRUVICA + BR y en el 2% de los pacientes tratados con placebo + BR. La frecuencia de fibrilación auricular de Grado 3 y 4 fue del 3% en pacientes tratados con IMBRUVICA + BR y del 1% en pacientes tratados con placebo + BR.
iLLUMINATE
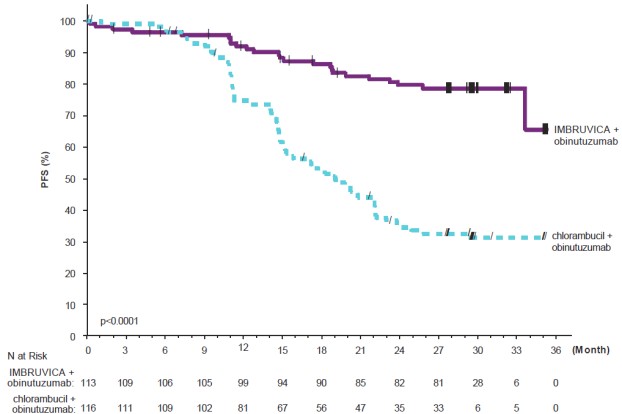
Las reacciones adversas descritas a continuación en la Tabla 12 reflejan la exposición a IMBRUVICA + obinutuzumab con una duración media de 29,3 meses y la exposición a clorambucilo + obinutuzumab con una mediana de 5,1 meses en iLLUMINATE en pacientes con CLL/SLL no tratados previamente.
| Sistema del Cuerpo Reacción Adversa |
IMBRUVICA + Obinutuzumab (N=113) |
Clorambucilo + Obinutuzumab (N=115) |
||
| Todos los Grados (%) |
Grado 3 o Superior (%) | Todos los Grados (%) |
Grado 3 o Superior (%) | |
| Trastornos de la sangre y del sistema linfático | ||||
| Neutropenia* | 48 | 39 | 64 | 48 |
| Trombocitopenia* | 36 | 19 | 28 | 11 |
| Anemia | 17 | 4 | 25 | 8 |
| Trastornos de la piel y del tejido subcutáneo | ||||
| Erupción cutánea* | 36 | 3 | 11 | 0 |
| Hematomas* | 32 | 3 | 3 | 0 |
| Trastornos gastrointestinales | ||||
| Diarrea | 34 | 3 | 10 | 0 |
| Estreñimiento | 16 | 0 | 12 | 1 |
| Náuseas | 12 | 0 | 30 | 0 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | ||||
| Dolor musculoesquelético* | 33 | 1 | 23 | 3 |
| Artralgia | 22 | 1 | 10 | 0 |
| Espasmos musculares | 13 | 0 | 6 | 0 |
| Trastornos respiratorios, torácicos y mediastínicos | ||||
| Tos | 27 | 1 | 12 | 0 |
| Lesiones, intoxicaciones y complicaciones de procedimientos terapéuticos | ||||
| Infusion related reaction | 25 | 2 | 58 | 8 |
| Trastornos vasculares | ||||
| Hemorrhage* | 25 | 1 | 9 | 0 |
| Hypertension* | 17 | 4 | 4 | 3 |
| Trastornos generales y alteraciones en el lugar de administración | ||||
| Pyrexia | 19 | 2 | 26 | 1 |
| Fatiga | 18 | 0 | 17 | 2 |
| Edema periférico | 12 | 0 | 7 | 0 |
| Infecciones e infestaciones | ||||
| Pneumonia* | 16 | 9 | 9 | 4† |
| Infección de las vías respiratorias superiores |
14 | 1 | 6 | 0 |
| Skin infection* | 13 | 1 | 3 | 0 |
| Infección del tracto urinario | 12 | 3 | 7 | 1 |
| Nasopharyngitis | 12 | 0 | 3 | 0 |
| Conjuntivitis | 11 | 0 | 2 | 0 |
| Trastornos del metabolismo y de la nutrición | ||||
| Hyperuricemia | 13 | 1 | 0 | 0 |
| Trastornos cardíacos | ||||
| Atrial fibrillation | 12 | 5 | 0 | 0 |
| Trastornos psiquiátricos | ||||
| Insomnio | 12 | 0 | 4 | 0 |
El sistema del cuerpo y los términos individuales de las reacciones adversas al medicamento (RAM) se clasifican en orden descendente de frecuencia en el grupo de IMBRUVICA.
* Incluye múltiples términos de RAM.
† Incluye un evento con desenlace fatal.
E1912
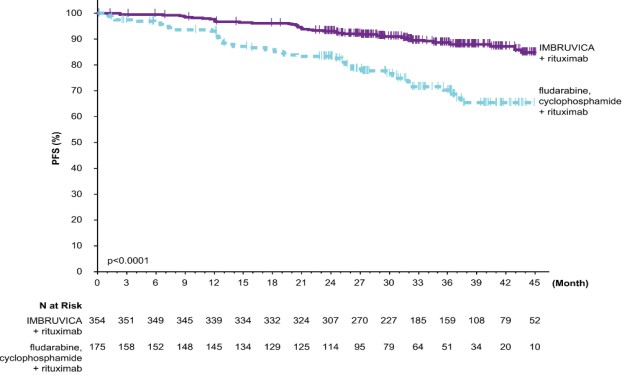
Las reacciones adversas descritas a continuación en la Tabla 13 reflejan la exposición a IMBRUVICA + rituximab con una duración media de 34,3 meses y la exposición a FCR con una mediana de 4,7 meses en el estudio E1912 en pacientes con CLL/SLL no tratados previamente que tenían 70 años o menos.
| Sistema del cuerpo Reacción adversa |
IMBRUVICA + Rituximab
(N=352) |
Fludarabina + Ciclofosfamida + Rituximab (N=158) |
||
| Todos los grados (%) |
Grado 3 o Superior (%) |
Todos los grados (%) |
Grado 3 o Superior (%) |
|
| Trastornos generales y alteraciones en el lugar de administración | ||||
| Fatiga | 80 | 2 | 78 | 3 |
| Edema periférico | 28 | 1 | 17 | 0 |
| Pirexia | 27 | 1 | 27 | 1 |
| Dolor | 23 | 2 | 8 | 0 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | ||||
| Dolor musculoesquelético* | 61 | 5 | 35 | 2 |
| Artralgia | 41 | 5 | 10 | 1 |
| Trastornos gastrointestinales | ||||
| Diarrea | 53 | 4 | 27 | 1 |
| Náuseas | 40 | 1 | 64 | 1 |
| Estomatitis* | 22 | 1 | 8 | 1 |
| Dolor abdominal* | 19 | 2 | 10 | 1 |
| Vómitos | 18 | 2 | 28 | 0 |
| Estreñimiento | 17 | 0 | 32 | 0 |
| Trastornos de la piel y del tejido subcutáneo | ||||
| Erupción cutánea* | 49 | 4 | 29 | 5 |
| Moretones* | 36 | 1 | 4 | 1 |
| Trastornos vasculares | ||||
| Hipertensión* | 42 | 19 | 22 | 6 |
| Hemorragia* | 31 | 2 | 8 | 1 |
| Trastornos del sistema nervioso | ||||
| Dolor de cabeza | 40 | 1 | 27 | 1 |
| Mareos | 21 | 1 | 13 | 1 |
| Peripheral neuropathy* | 19 | 1 | 13 | 1 |
| Trastornos respiratorios, torácicos y mediastínicos | ||||
| Tos | 32 | 0 | 25 | 0 |
| Disnea | 22 | 2 | 21 | 1 |
| Infecciones e infestaciones | ||||
| Infección de las vías | 29 | 1 | 19 | 2 |
| respiratorias superiores | ||||
| Skin infection* | 16 | 1 | 3 | 1 |
| Trastornos del metabolismo y la nutrición | ||||
| Hiperuricemia | 19 | 1 | 4 | 0 |
| Disminución del apetito | 15 | 0 | 20 | 1 |
| Trastornos psiquiátricos | ||||
| Insomnio | 16 | 1 | 19 | 1 |
El sistema del cuerpo y los términos individuales de las reacciones adversas a medicamentos (RAM) se ordenan en orden descendente de frecuencia en el grupo de IMBRUVICA.
* Incluye múltiples términos de RAM.
| IMBRUVICA + Rituximab
(N=352) |
Fludarabina + Ciclofosfamida + Rituximab (N=158) |
|||
| Todos los Grados (%) |
Grado 3 o 4 (%) |
Todos los Grados (%) |
Grado 3 o 4 (%) |
|
| Anormalidades hematológicas Neutrófilos disminuidos Plaquetas disminuidas Hemoglobina disminuida |
53 43 26 |
30 7 0 |
70 69 51 |
44 25 2 |
| Anormalidades químicas Creatinina aumentada Bilirrubina aumentada AST aumentada |
38 30 25 |
1 2 3 |
17 15 23 |
1 0 <1 |
Basado en mediciones de laboratorio según los criterios del IWCLL.
Macroglobulinemia de Waldenström
Los datos que se describen a continuación reflejan la exposición a IMBRUVICA en dos ensayos clínicos de un solo brazo (Estudio 1118 y el brazo de monoterapia de INNOVATE) y un ensayo controlado aleatorizado (INNOVATE), que incluyeron un total de 169 pacientes con WM expuestos a IMBRUVICA. El estudio 1118 incluyó a 63 pacientes con WM previamente tratada que recibieron IMBRUVICA como agente único. INNOVATE incluyó a 150 pacientes con WM sin tratamiento previo o previamente tratada que recibieron IMBRUVICA o placebo en combinación con rituximab. El brazo de monoterapia de INNOVATE incluyó a 31 pacientes con WM previamente tratada que recibieron IMBRUVICA después del fracaso de la terapia previa con rituximab.
Las reacciones adversas más comunes en los estudios 1118 e INNOVATE (≥ 20%) fueron neutropenia, diarrea, hematomas, trombocitopenia, hemorragia, dolor musculoesquelético, erupción cutánea y náuseas.
El cinco por ciento de los pacientes que recibieron IMBRUVICA en los estudios 1118 e INNOVATE interrumpieron el tratamiento debido a reacciones adversas. La reacción adversa más común que condujo a la interrupción fue la fibrilación auricular. Se produjeron reacciones adversas que condujeron a la reducción de la dosis en el 14% de los pacientes.
Estudio 1118 y brazo de monoterapia de INNOVATE
Las reacciones adversas y las anomalías de laboratorio que se describen a continuación en la Tabla 15 y la Tabla 16 reflejan la exposición a IMBRUVICA con una duración media de 11,7 meses en el Estudio 1118 y 33 meses en el brazo de monoterapia de INNOVATE.
| Sistema del cuerpo | Reacción adversa | Todos los grados (%) | Grado 3 o Superior (%) |
| Trastornos gastrointestinales | Diarrea Náuseas Estomatitis* Estreñimiento Enfermedad por reflujo gastroesofágico |
38 21 15 12 12 |
2 0 0 1 0 |
| Trastornos de la piel y del tejido subcutáneo | Hematomas* Erupción cutánea* |
28 21 |
1 1 |
| Trastornos vasculares | Hemorragia* Hipertensión* |
28 14 |
0 4 |
| Trastornos generales y condiciones del lugar de administración | Fatiga Pirexia |
18 12 |
2 2 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | Dolor musculoesquelético* Espasmos musculares |
21 19 |
0 0 |
| Infecciones e infestaciones | Infección de las vías respiratorias superiores Infección de la piel* Sinusitis* Neumonía* |
19 18 16 13 |
0 3 0 5 |
| Trastornos del sistema nervioso | Dolor de cabeza Mareos |
14 13 |
0 0 |
| Trastornos respiratorios, torácicos y mediastínicos | Tos | 13 | 0 |
El sistema del cuerpo y los términos preferidos de las reacciones adversas al medicamento (RAM) individuales se ordenan en orden descendente de frecuencia.
* Incluye múltiples términos de RAM.
| Porcentaje de pacientes (N=94) | ||
| Todos los grados (%) | Grado 3 o 4 (%) | |
| Disminución de plaquetas | 38 | 11 |
| Disminución de neutrófilos | 43 | 16 |
| Disminución de hemoglobina | 21 | 6 |
Se produjo trombocitopenia de grado 4 emergente del tratamiento (4%) y neutropenia (7%) en los pacientes.
INNOVATE
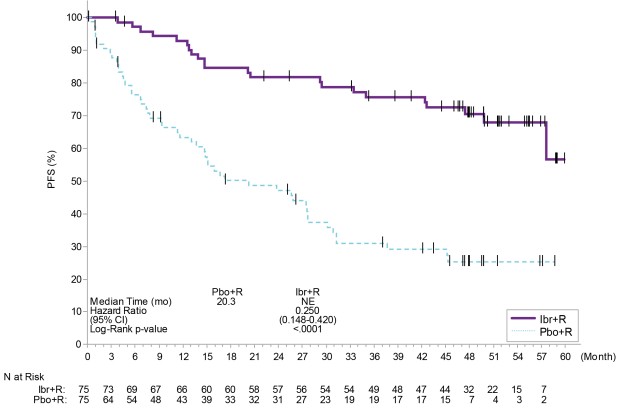
Las reacciones adversas descritas a continuación en la Tabla 17 reflejan la exposición a IMBRUVICA + R con una duración media de 25,8 meses y la exposición a placebo + R con una duración media de 15,5 meses en pacientes con WM sin tratamiento previo o previamente tratados en INNOVATE.
| Sistema del cuerpo Reacción adversa |
IMBRUVICA + R (N=75) |
Placebo + R (N=75) |
||
| Todos los grados (%) |
Grado 3 o superior (%) |
Todos los grados (%) |
Grado 3 o superior (%) |
|
| Trastornos de la piel y del tejido subcutáneo | ||||
| Moretones* | 37 | 1 | 5 | 0 |
| Erupción cutánea* | 24 | 1 | 11 | 0 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | ||||
| Dolor musculoesquelético* | 35 | 4 | 21 | 3 |
| Artralgia | 24 | 3 | 11 | 1 |
| Espasmos musculares | 17 | 0 | 12 | 1 |
| Trastornos vasculares | ||||
| Hemorragia* | 32 | 3 | 17 | 4† |
| Hipertensión* | 20 | 13 | 5 | 4 |
| Trastornos gastrointestinales | ||||
| Diarrea | 28 | 0 | 15 | 1 |
| Náuseas | 21 | 0 | 12 | 0 |
| Dispepsia | 16 | 0 | 1 | 0 |
| Estreñimiento | 13 | 1 | 11 | 1 |
| Infecciones e infestaciones | ||||
| Neumonía* | 19 | 13 | 5 | 3 |
| Infección de la piel* | 17 | 3 | 3 | 0 |
| Infección del tracto urinario | 13 | 0 | 0 | 0 |
| Bronquitis | 12 | 3 | 7 | 0 |
| Influenza/Gripe | 12 | 0 | 7 | 1 |
| Infección viral de las vías respiratorias superiores | 11 | 0 | 7 | 0 |
| Trastornos generales y afecciones en el sitio de administración | ||||
| Edema periférico | 17 | 0 | 12 | 1 |
| Trastornos respiratorios, torácicos y mediastínicos | ||||
| Tos | 17 | 0 | 11 | 0 |
| Trastornos de la sangre y del sistema linfático | ||||
| Neutropenia* | 16 | 12 | 11 | 4 |
| Trastornos cardíacos | ||||
| Fibrilación auricular | 15 | 12 | 3 | 1 |
| Trastornos del sistema nervioso | ||||
| Mareo | 11 | 0 | 7 | 0 |
| Trastornos psiquiátricos | ||||
| Insomnio | 11 | 0 | 4 | 0 |
| Trastornos del metabolismo y de la nutrición | ||||
| Hipopotasemia/Hipocaliemia | 11 | 0 | 1 | 1 |
El sistema del cuerpo y los términos preferidos de reacciones adversas a medicamentos individuales se ordenan en orden descendente de frecuencia.
* Incluye múltiples términos de reacciones adversas a medicamentos.
† Incluye un evento con resultado fatal.
Se observaron reacciones relacionadas con la infusión de Grado 3 o 4 en el 1% de los pacientes tratados con IR.
Enfermedad crónica de injerto contra huésped
Estudio 1129
Los datos que se describen a continuación reflejan la exposición a IMBRUVICA en un ensayo clínico abierto (Estudio 1129) que incluyó a 42 pacientes con EICHc después del fracaso de la terapia con corticosteroides de primera línea y requirieron terapia adicional [ver Estudios Clínicos (14.3)].
Las reacciones adversas más comunes en el Estudio 1129 (≥ 20%) fueron fatiga, hematomas, diarrea, trombocitopenia, estomatitis, espasmos musculares, náuseas, hemorragia, anemia y neumonía. La fibrilación auricular ocurrió en un paciente (2%) que fue de Grado 3.
El veinticuatro por ciento de los pacientes que recibieron IMBRUVICA en el Estudio 1129 suspendieron el tratamiento debido a reacciones adversas. Las reacciones adversas más comunes que llevaron a la suspensión fueron fatiga y neumonía. Las reacciones adversas que llevaron a la reducción de la dosis ocurrieron en el 26% de los pacientes.
Las reacciones adversas y las anomalías de laboratorio que se describen a continuación en la Tabla 18 y la Tabla 19 reflejan la exposición a IMBRUVICA con una duración media de 4,4 meses en el Estudio 1129.
| Sistema del cuerpo | Reacción adversa | Todos los grados (%) | Grado 3 o superior (%) |
| Trastornos generales y afecciones en el sitio de administración | Fatiga Pirexia Edema periférico |
57 17 12 |
12 5 0 |
| Trastornos de la piel y del tejido subcutáneo | Hematomas* Erupción cutánea* |
40 12 |
0 0 |
| Trastornos gastrointestinales | Diarrea Estomatitis* Náuseas Estreñimiento |
36 29 26 12 |
10 2 0 0 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | Espasmos musculares Dolor musculoesquelético* |
29 14 |
2 5 |
| Trastornos vasculares | Hemorragia* | 26 | 0 |
| Infecciones e infestaciones | Neumonía* Infección de las vías respiratorias superiores Sepsis* |
21 19 10 |
14† 0 10 |
| Trastornos del sistema nervioso | Dolor de cabeza | 17 | 5 |
| Lesiones, intoxicaciones y complicaciones de procedimientos | Caída | 17 | 0 |
| Trastornos respiratorios, torácicos y mediastínicos | Tos Disnea |
14 12 |
0 2 |
| Trastornos del metabolismo y la nutrición | Hipopotasemia | 12 | 7 |
El sistema de clasificación de órganos y los términos preferidos de reacciones adversas al medicamento (RAM) individuales se ordenan en orden descendente de frecuencia.
* Incluye múltiples términos de RAM.
† Incluye 2 eventos con desenlace fatal.
| Porcentaje de Pacientes (N=42) | ||
| Todos los Grados (%) | Grado 3 o 4 (%) | |
| Platelets decreased | 33 | 0 |
| Neutrophils decreased | 10 | 10 |
| Hemoglobin decreased | 24 | 2 |
La neutropenia de Grado 4 emergente del tratamiento ocurrió en el 2% de los pacientes.
iMAGINE
La seguridad de IMBRUVICA se evaluó en el estudio iMAGINE, que incluyó a 47 pacientes pediátricos y adultos jóvenes de 1 año a menos de 22 años de edad con cGVHD después del fracaso de una o más líneas de terapia sistémica. Los pacientes de 12 años o mayores fueron tratados con IMBRUVICA 420 mg por vía oral una vez al día, y los pacientes de 1 año a menos de 12 años fueron tratados con IMBRUVICA 240 mg/m2 por vía oral una vez al día [ver Estudios Clínicos (14.3)]. La mediana de la duración de la exposición a IMBRUVICA fue de 7.1 meses (rango, 0.2 a 25.9 meses).
Las reacciones adversas graves ocurrieron en el 64% de los pacientes que recibieron IMBRUVICA. Las reacciones adversas graves en más de dos pacientes incluyeron neumonía, pirexia, sepsis y estomatitis. Las reacciones adversas fatales ocurrieron en dos pacientes que recibieron IMBRUVICA, incluyendo sepsis y síndrome de dificultad respiratoria aguda (SDRA).
La interrupción permanente de IMBRUVICA debido a una reacción adversa ocurrió en el 23% de los pacientes. Las reacciones adversas que resultaron en la interrupción permanente en al menos dos pacientes incluyeron hemorragia. Las reducciones de dosis de IMBRUVICA debido a una reacción adversa ocurrieron en el 19% de los pacientes. Las reacciones adversas que requirieron reducción de la dosis en al menos dos pacientes incluyeron estomatitis.
Las reacciones adversas más comunes (≥ 20%), incluyendo anomalías de laboratorio, fueron anemia, dolor musculoesquelético, pirexia, diarrea, neumonía, dolor abdominal, estomatitis, trombocitopenia y dolor de cabeza.
Tabla 20 resume las reacciones adversas en iMAGINE.
| IMBRUVICA (N=47) |
||
| Sistema Corporal Reacción Adversa |
Todos los Grados (%) |
Grado 3 o 4 (%) |
| Trastornos generales y afecciones en el lugar de administración | ||
| Pirexia | 30 | 11 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | ||
| Dolor musculoesquelético* | 30 | 2 |
| Osteonecrosis | 11 | 9 |
| Trastornos gastrointestinales | ||
| Diarrea | 28 | 2 |
| Dolor abdominal* | 23 | 4 |
| Estomatitis* | 23 | 9 |
| Vómitos | 19 | 2 |
| Náuseas | 19 | 4 |
| Infecciones e infestaciones | ||
| Neumonía* | 23 | 13 |
| Infección de la piel* | 17 | 4 |
| Sepsis* | 11 | 9† |
| Trastornos del sistema nervioso | ||
| Dolor de cabeza | 21 | 2 |
| Trastornos de la piel y del tejido subcutáneo | ||
| Erupción cutánea* | 19 | 2 |
| Prurito | 13 | 0 |
| Petequias | 13 | 0 |
| Trastornos respiratorios, torácicos y mediastínicos | ||
| Tos | 19 | 2 |
| Trastornos vasculares | ||
| Hemorrhage* | 17 | 0 |
| Hipertensión* | 11 | 4 |
| Trastornos hematológicos y del sistema linfático | ||
| Hipopotasemia | 15 | 6 |
| Hypogammaglobulinemia* | 11 | 0 |
| Trastornos cardíacos | ||
| Taquicardia sinusal | 11 | 0 |
| Investigaciones | ||
| Alanine aminotransferase increased | 11 | 2 |
La clase de sistema de órganos y los términos preferidos de reacciones adversas a medicamentos (RAM) individuales se ordenan en orden descendente de frecuencia.
* Incluye múltiples términos de RAM.
† Incluye 1 desenlace fatal.
Tabla 21 resume las anomalías de laboratorio en iMAGINE.
| IMBRUVICA (N=47) |
||
| Todos los grados (%) |
Grado 3 o 4 (%) |
|
| Hemoglobin decreased | 49 | 13 |
| Platelets decreased | 21 | 4 |
| Neutrophils decreased | 13 | 6 |
La neutropenia de Grado 4 emergente del tratamiento ocurrió en el 3% de los pacientes.
Reacciones Adversas Importantes Adicionales
Eventos Cardiovasculares
Los datos sobre eventos cardiovasculares se basan en ensayos controlados aleatorios con IMBRUVICA (n=2115; duración media del tratamiento de 19,1 meses para 1157 pacientes tratados con IMBRUVICA y 5,3 meses para 958 pacientes en el grupo de control). La incidencia de taquiarritmias ventriculares (extrasístoles ventriculares, arritmias ventriculares, fibrilación ventricular, aleteo ventricular y taquicardia ventricular) de cualquier grado fue del 1,0% frente al 0,4% y de Grado 3 o superior fue del 0,3% frente al 0% en pacientes tratados con IMBRUVICA en comparación con los pacientes del grupo de control. La incidencia de fibrilación auricular y aleteo auricular de cualquier grado fue del 8,4% frente al 1,6% y para Grado 3 o superior fue del 4,0% frente al 0,5% en pacientes tratados con IMBRUVICA en comparación con los pacientes del grupo de control. Además, la incidencia de insuficiencia cardíaca de cualquier grado fue del 1,7% frente al 0,5% y para Grado 3 o superior fue del 1,2% frente al 0,3% en pacientes tratados con IMBRUVICA en comparación con los pacientes del grupo de control.
La incidencia de eventos cerebrovasculares isquémicos (accidentes cerebrovasculares, accidente cerebrovascular isquémico, isquemia cerebral y ataque isquémico transitorio) de cualquier grado fue del 1% frente al 0,4% y de Grado 3 o superior fue del 0,5% frente al 0,2% en pacientes tratados con IMBRUVICA en comparación con los pacientes del grupo de control, respectivamente.
Diarrea
En ensayos controlados aleatorios (n=2115; duración media del tratamiento de 19,1 meses para 1157 pacientes tratados con IMBRUVICA y 5,3 meses para 958 pacientes en el grupo de control), la diarrea de cualquier grado se produjo a una tasa del 43% de los pacientes tratados con IMBRUVICA en comparación con el 19% de los pacientes en el grupo de control. La diarrea de Grado 3 se produjo en el 3% frente al 1% de los pacientes tratados con IMBRUVICA en comparación con el grupo de control, respectivamente. Menos del 1% (0,3%) de los sujetos interrumpieron el tratamiento con IMBRUVICA debido a la diarrea en comparación con el 0% en el grupo de control.
Según los datos de 1605 de estos pacientes, la mediana del tiempo hasta la primera aparición fue de 21 días (rango, 0 a 708) frente a 46 días (rango, 0 a 492) para la diarrea de cualquier grado y 117 días (rango, 3 a 414) frente a 194 días (rango, 11 a 325) para la diarrea de Grado 3 en pacientes tratados con IMBRUVICA en comparación con el grupo de control, respectivamente. De los pacientes que informaron diarrea, el 85% frente al 89% tuvieron una resolución completa, y el 15% frente al 11% no habían informado de resolución en el momento del análisis en los pacientes tratados con IMBRUVICA en comparación con el grupo de control, respectivamente. La mediana del tiempo desde el inicio hasta la resolución en los sujetos tratados con IMBRUVICA fue de 7 días (rango, 1 a 655) frente a 4 días (rango, 1 a 367) para la diarrea de cualquier grado y 7 días (rango, 1 a 78) frente a 19 días (rango, 1 a 56) para la diarrea de Grado 3 en los sujetos tratados con IMBRUVICA en comparación con el grupo de control, respectivamente.
Trastorno Visual
En ensayos controlados aleatorios (n=2115; duración media del tratamiento de 19,1 meses para 1157 pacientes tratados con IMBRUVICA y 5,3 meses para 958 pacientes en el grupo de control), la visión borrosa y la disminución de la agudeza visual de cualquier grado se produjeron en el 11% de los pacientes tratados con IMBRUVICA (9% Grado 1, 2% Grado 2, ningún Grado 3 o superior) en comparación con el 6% en el grupo de control (5% Grado 1 y < 1% Grado 2 y 3).
Según los datos de 1605 de estos pacientes, la mediana del tiempo hasta la primera aparición fue de 91 días (rango, 0 a 617) frente a 100 días (rango, 2 a 477) en los pacientes tratados con IMBRUVICA en comparación con el grupo de control, respectivamente. De los pacientes que informaron trastornos visuales, el 60% frente al 71% tuvieron una resolución completa y el 40% frente al 29% no habían informado de resolución en el momento del análisis en los pacientes tratados con IMBRUVICA en comparación con el grupo de control, respectivamente. La mediana del tiempo desde el inicio hasta la resolución fue de 37 días (rango, 1 a 457) frente a 26 días (rango, 1 a 721) en los sujetos tratados con IMBRUVICA en comparación con el grupo de control, respectivamente.
6.2
Experiencia Poscomercialización
Las siguientes reacciones adversas se han identificado durante el uso poscomercialización de IMBRUVICA. Debido a que estas reacciones se notifican voluntariamente a partir de una población de tamaño incierto, no siempre es posible estimar de forma fiable su frecuencia o establecer una relación causal con la exposición al fármaco.
- Trastornos hepatobiliares: insuficiencia hepática, incluyendo eventos agudos y/o mortales, cirrosis hepática, lesión hepática inducida por fármacos
- Trastornos respiratorios: enfermedad pulmonar intersticial
- Trastornos del metabolismo y la nutrición: síndrome de lisis tumoral
- Trastornos del sistema inmunitario: shock anafiláctico, angioedema, urticaria
- Trastornos de la piel y del tejido subcutáneo: Síndrome de Stevens-Johnson (SSJ), onicoclasis, paniculitis, dermatosis neutrofílicas, vasculitis cutánea
- Infecciones: reactivación de la hepatitis B
- Trastornos del sistema nervioso: neuropatía periférica
7 INTERACCIONES MEDICAMENTOSAS
7.1
Efecto de los inhibidores de CYP3A sobre Ibrutinib
La administración conjunta de IMBRUVICA con un inhibidor potente o moderado de CYP3A puede aumentar las concentraciones plasmáticas de ibrutinib [ver Farmacología clínica (12.3)]. El aumento de las concentraciones de ibrutinib puede aumentar el riesgo de toxicidad relacionada con el fármaco.
Se recomiendan modificaciones de la dosis de IMBRUVICA cuando se usa concomitantemente con posaconazol, voriconazol e inhibidores moderados de CYP3A [ver Posología y administración (2.3)].
Evite el uso concomitante de otros inhibidores potentes de CYP3A. Interrumpa el tratamiento con IMBRUVICA si se van a utilizar estos inhibidores a corto plazo (como antiinfecciosos durante siete días o menos) [ver Posología y administración (2.3)].
Evite el pomelo y las naranjas amargas durante el tratamiento con IMBRUVICA, ya que estos contienen inhibidores potentes o moderados de CYP3A.
7.2
Efecto de los inductores de CYP3A sobre Ibrutinib
La administración conjunta de IMBRUVICA con inductores potentes de CYP3A puede disminuir las concentraciones de ibrutinib. Evite la administración conjunta con inductores potentes de CYP3A [ver Farmacología clínica (12.3)].
8 USO EN POBLACIONES ESPECÍFICAS
8.1
Embarazo
Resumen de Riesgo
IMBRUVICA puede causar daño fetal según los hallazgos de estudios en animales. No hay datos disponibles sobre el uso de IMBRUVICA en mujeres embarazadas para informar sobre un riesgo asociado con el fármaco de defectos congénitos importantes y aborto espontáneo. En estudios de reproducción en animales, la administración de ibrutinib a ratas y conejas embarazadas durante el período de organogénesis con exposiciones de hasta 3 a 20 veces la dosis clínica de 420 mg diarios produjo toxicidad embriofetal, incluidas anomalías estructurales (ver Datos). Advertir a las mujeres embarazadas sobre el riesgo potencial para el feto.
Todos los embarazos tienen un riesgo de fondo de defectos de nacimiento, pérdida u otros resultados adversos. El riesgo de fondo estimado de defectos congénitos importantes y aborto espontáneo para la población indicada es desconocido. En la población general de EE. UU., el riesgo de fondo estimado de defectos congénitos importantes y aborto espontáneo en embarazos clínicamente reconocidos es del 2-4 % y del 15-20 %, respectivamente.
Datos
Datos en animales
El ibrutinib se administró por vía oral a ratas embarazadas durante el período de organogénesis a dosis de 10, 40 y 80 mg/kg/día. El ibrutinib a una dosis de 80 mg/kg/día se asoció con malformaciones viscerales (corazón y vasos sanguíneos principales) y un aumento de las reabsorciones y la pérdida postimplantacional. La dosis de 80 mg/kg/día en ratas es aproximadamente 20 veces la exposición en pacientes con LMC/LMC de células B pequeñas o WM a los que se les administra una dosis de 420 mg diarios. El ibrutinib a dosis de 40 mg/kg/día o más se asoció con una disminución del peso fetal. La dosis de 40 mg/kg/día en ratas es aproximadamente 8 veces la exposición (AUC) en pacientes a los que se les administra una dosis de 420 mg diarios.
El ibrutinib también se administró por vía oral a conejas embarazadas durante el período de organogénesis a dosis de 5, 15 y 45 mg/kg/día. El ibrutinib a una dosis de 15 mg/kg/día o más se asoció con variaciones esqueléticas (esternón fusionado) y el ibrutinib a una dosis de 45 mg/kg/día se asoció con un aumento de las reabsorciones y la pérdida postimplantacional. La dosis de 15 mg/kg/día en conejos es aproximadamente 2,8 veces la exposición en pacientes con LMC/LMC de células B pequeñas o WM a los que se les administra una dosis de 420 mg diarios.
8.2
Lactancia
Resumen de Riesgo
No hay información sobre la presencia de ibrutinib o sus metabolitos en la leche materna, los efectos en el niño amamantado o los efectos en la producción de leche. Debido al potencial de reacciones adversas graves en el niño amamantado, se debe aconsejar a las mujeres que no amamanten durante el tratamiento con IMBRUVICA y durante 1 semana después de la última dosis.
8.3
Mujeres y hombres en edad fértil
IMBRUVICA puede causar daño fetal cuando se administra a mujeres embarazadas [ver Uso en poblaciones específicas (8.1)].
Prueba de embarazo
Verificar el estado del embarazo en mujeres en edad fértil antes de iniciar el tratamiento con IMBRUVICA.
Anticoncepción
Mujeres
Se debe aconsejar a las mujeres en edad fértil que utilicen métodos anticonceptivos eficaces durante el tratamiento con IMBRUVICA y durante 1 mes después de la última dosis.
Hombres
Se debe aconsejar a los hombres con parejas femeninas en edad fértil que utilicen métodos anticonceptivos eficaces durante el tratamiento con IMBRUVICA y durante 1 mes después de la última dosis.
8.4
Uso pediátrico
Enfermedad injerto contra huésped (EICH) crónica
La seguridad y la eficacia de IMBRUVICA se han establecido para el tratamiento de la EICH crónica después del fracaso de una o más líneas de terapia sistémica en pacientes pediátricos de 1 año de edad o más.
El uso de IMBRUVICA para esta indicación se basa en la evidencia del estudio iMAGINE, que incluyó pacientes pediátricos de 1 año de edad o más con EICH previamente tratada, incluidos pacientes en los siguientes grupos de edad: un paciente de 1 año a menos de 2 años de edad, 20 pacientes de 2 años a menos de 12 años de edad y 19 pacientes de 12 años a menos de 17 años de edad. Se proporcionaron datos adicionales de eficacia de apoyo del Estudio 1129 en adultos [ver Reacciones adversas (6.1), Farmacología clínica (12.3) y Estudios clínicos (14.3)].
La dosis recomendada de IMBRUVICA en pacientes de 12 años o más es la misma que en adultos, y la dosis recomendada en pacientes de 1 año a menos de 12 años de edad se basa en el área de superficie corporal (ASC) [ver Posología y administración (2.1)].
La seguridad y la eficacia de IMBRUVICA no se han establecido para esta indicación en pacientes pediátricos menores de 1 año de edad.
Linfoma no Hodgkin de células B maduras
Se evaluó la seguridad y la eficacia de IMBRUVICA en combinación con quimioinmunoterapia, pero no se han establecido en base a un estudio aleatorizado abierto (NCT02703272) en 35 pacientes, que incluyó 26 pacientes pediátricos de 5 a menos de 17 años, con linfoma no Hodgkin de células B maduras previamente tratado. El estudio se interrumpió por falta de eficacia. En la población aleatorizada, la hemorragia mayor y la interrupción de la quimioinmunoterapia debido a reacciones adversas ocurrieron con mayor frecuencia en el brazo de ibrutinib más quimioinmunoterapia en comparación con el brazo de quimioinmunoterapia sola.
LMC/LMC de células B pequeñas, LMC/LMC de células B pequeñas con deleción 17p, WM
La seguridad y la eficacia de IMBRUVICA en pacientes pediátricos no se han establecido en LMC/LMC de células B pequeñas, LMC/LMC de células B pequeñas con deleción 17p o WM.
![La siguiente estructura para el Ibrutinib es un inhibidor de la quinasa. Es un sólido blanco a blanquecino con la fórmula empírica C25H24N6O2 y un peso molecular de 440,50. El ibrutinib es libremente soluble en dimetilsulfóxido, soluble en metanol y prácticamente insoluble en agua. El nombre químico del ibrutinib es 1-[(3R)-3-[4-amino-3-(4-fenoxifenil)-1H-pirazolo[3,4 d]pirimidin-1-il]-1-piperidinil]-2-propen-1-ona y tiene](/images/44/imbruvica-01.jpg)