
Fabricante de medicamentos: Bayer HealthCare Pharmaceuticals Inc. (Updated: 2020-12-09)
DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
STIVARGA® (regorafenib) comprimidos recubiertos con película, para uso oral
Aprobación inicial en EE. UU.: 2012
ADVERTENCIA: HEPATOTOXICIDAD
Consulte la información de prescripción completa para ver la advertencia completa en el recuadro.
- •
- En ensayos clínicos se ha producido hepatotoxicidad grave y a veces mortal. (5.1)
- •
- Controle la función hepática antes y durante el tratamiento. (5.1)
- •
- Interrumpa y luego reduzca o suspenda STIVARGA por hepatotoxicidad manifestada por pruebas elevadas de la función hepática o necrosis hepatocelular, dependiendo de la gravedad y la persistencia. (2.2)
INDICACIONES Y USO
STIVARGA es un inhibidor de la quinasa indicado para el tratamiento de pacientes con:
- •
- Cáncer colorrectal metastásico (CCRm) que han sido tratados previamente con quimioterapia basada en fluoropirimidina, oxaliplatino e irinotecán, una terapia anti-VEGF y, si el RAS es de tipo salvaje, una terapia anti-EGFR. (1.1)
- •
- Tumor del estroma gastrointestinal (GIST) localmente avanzado, irresecable o metastásico que han sido tratados previamente con mesilato de imatinib y malato de sunitinib. (1.2)
- •
- Carcinoma hepatocelular (CHC) que han sido tratados previamente con sorafenib (1.3)
POSOLOGÍA Y ADMINISTRACIÓN
FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Comprimidos: 40 mg (3)
CONTRAINDICACIONES
Ninguna. (4)
ADVERTENCIAS Y PRECAUCIONES
- •
- Hepatotoxicidad: Controle las pruebas de función hepática. Suspenda y luego reduzca o suspenda STIVARGA según la gravedad y la duración. (5.1)
- •
- Infecciones: Suspenda STIVARGA en pacientes con infecciones graves o que empeoran. (5.2)
- •
- Hemorragia: Suspenda permanentemente STIVARGA por hemorragia grave o potencialmente mortal. (5.3)
- •
- Perforación o fístula gastrointestinal: Suspenda STIVARGA. (5.4)
- •
- Toxicidad dermatológica: Suspenda y luego reduzca o suspenda STIVARGA según la gravedad y la persistencia de la toxicidad dermatológica. (5.5)
- •
- Hipertensión: Suspenda temporal o permanentemente STIVARGA por hipertensión grave o no controlada. (5.6)
- •
- Isquemia e infarto cardíaco: Suspenda STIVARGA por nueva isquemia/infarto cardíaco agudo y reanude solo después de la resolución de los eventos isquémicos agudos. (5.7)
- •
- Síndrome de leucoencefalopatía posterior reversible (SLPR): Suspenda STIVARGA. (5.8)
- •
- Riesgo de cicatrización de heridas alterada: Suspenda durante al menos 2 semanas antes de la cirugía electiva. No administre durante al menos 2 semanas después de una cirugía mayor y hasta que la herida cicatrice adecuadamente. No se ha establecido la seguridad de reanudar STIVARGA después de la resolución de las complicaciones de la cicatrización de la herida. (5.9)
- •
- Toxicidad embriofetal: Puede causar daño fetal. Informe a las mujeres sobre el riesgo potencial para el feto y que utilicen un método anticonceptivo eficaz durante el tratamiento y durante 2 meses después de la dosis final. Aconseje a los hombres que usen un método anticonceptivo eficaz durante 2 meses después de la dosis final. (5.10,8.1, 8.3)
REACCIONES ADVERSAS
Las reacciones adversas más frecuentes (≥20%) son dolor (incluido dolor gastrointestinal y abdominal), SEMP, astenia/fatiga, diarrea, disminución del apetito/ingesta de alimentos, hipertensión, infección, disfonía, hiperbilirrubinemia, fiebre, mucositis, pérdida de peso, erupción y náuseas. (6)
Para informar SOSPECHAS DE REACCIONES ADVERSAS, comuníquese con Bayer HealthCare Pharmaceuticals Inc. al 1-888-842-2937 o con la FDA al 1-800-FDA-1088 o en www.fda.gov/medwatch.
INTERACCIONES MEDICAMENTOSAS
- •
- Inductores potentes del CYP3A4: Evite los inductores potentes del CYP3A4. (7.1)
- •
- Inhibidores potentes del CYP3A4: Evite los inhibidores potentes del CYP3A4. (7.2)
- •
- Sustratos de BCRP: Controle de cerca a los pacientes para detectar síntomas de aumento de la exposición a los sustratos de BCRP. (7.3)
USO EN POBLACIONES ESPECÍFICAS
Madres lactantes: Suspenda el medicamento o la lactancia, teniendo en cuenta la importancia del medicamento para la madre. (8.3)
Consulte la sección 17 para obtener INFORMACIÓN DE ASESORAMIENTO PARA EL PACIENTE y el etiquetado para el paciente aprobado por la FDA.
Revisado: 12/2020
Tabla de Contenido
INFORMACIÓN DE PRESCRIPCIÓN COMPLETA: CONTENIDO*
ADVERTENCIA: HEPATOTOXICIDAD
1 INDICACIONES Y USO
1.1 Cáncer Colorrectal
1.2 Tumores del Estroma Gastrointestinal
1.3 Carcinoma Hepatocelular
2 DOSIS Y ADMINISTRACIÓN
2.1 Dosis Recomendada
2.2 Modificaciones de Dosis
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Hepatotoxicidad
5.2 Infecciones
5.3 Hemorragia
5.4 Perforación o Fístula Gastrointestinal
5.5 Toxicidad Dermatológica
5.6 Hipertensión
5.7 Isquemia e Infarto Cardíaco
5.8 Síndrome de Leucoencefalopatía Posterior Reversible
5.9 Riesgo de Deterioro en la Cicatrización de Heridas
5.10 Toxicidad Embrio-Fetal
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos
6.2 Experiencia Poscomercialización
7 INTERACCIONES MEDICAMENTOSAS
7.1 Efecto de Fuertes Inductores de CYP3A4 sobre Regorafenib
7.2 Efecto de Fuertes Inhibidores de CYP3A4 sobre Regorafenib
7.3 Efecto de Regorafenib sobre Sustratos de la Proteína de Resistencia al Cáncer de Mama (BCRP)
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y Hombres con Potencial Reproductivo
8.4 Uso Pediátrico
8.5 Uso Geriátrico
8.6 Insuficiencia Hepática
8.7 Insuficiencia Renal
8.8 Raza
10 SOBREDOSIS
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.2 Farmacodinámica
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
13.2 Toxicología Animal y/o Farmacología
14 ESTUDIOS CLÍNICOS
14.1 Cáncer Colorrectal
14.2 Tumores del Estroma Gastrointestinal
14.3 Carcinoma Hepatocelular (CHC)
16 PRESENTACIÓN/ALMACENAMIENTO Y MANIPULACIÓN
17 INFORMACIÓN DE ASESORAMIENTO PARA EL PACIENTE
- *
- No se enumeran las secciones o subsecciones omitidas de la información de prescripción completa.
ADVERTENCIA RECUADRADA
ADVERTENCIA: HEPATOTOXICIDAD
- •
- En ensayos clínicos ha ocurrido hepatotoxicidad grave y a veces mortal [ver Advertencias y precauciones (5.1)].
- •
- Monitorear la función hepática antes y durante el tratamiento [ver Advertencias y precauciones (5.1)].
- •
- Interrumpir y luego reducir o suspender STIVARGA por hepatotoxicidad manifestada por pruebas de función hepática elevadas o necrosis hepatocelular, dependiendo de la gravedad y persistencia [ver Dosificación y administración (2.2)].
1 INDICACIONES Y USO
1.1 Cáncer Colorrectal
STIVARGA está indicado para el tratamiento de pacientes con cáncer colorrectal (CCR) metastásico que han sido previamente tratados con quimioterapia basada en fluoropirimidina, oxaliplatino e irinotecán, una terapia anti-VEGF y, si es RAS de tipo salvaje, una terapia anti-EGFR.
1.2 Tumores del Estroma Gastrointestinal
STIVARGA está indicado para el tratamiento de pacientes con tumor del estroma gastrointestinal (GIST) localmente avanzado, irresecable o metastásico que han sido previamente tratados con imatinib mesilato y sunitinib malato.
1.3 Carcinoma Hepatocelular
STIVARGA está indicado para el tratamiento de pacientes con carcinoma hepatocelular (CHC) que han sido previamente tratados con sorafenib.
2 DOSIS Y ADMINISTRACIÓN
2.1 Dosis recomendada
La dosis recomendada es 160 mg de STIVARGA (cuatro comprimidos de 40 mg) por vía oral una vez al día durante los primeros 21 días de cada ciclo de 28 días. Continúe el tratamiento hasta la progresión de la enfermedad o toxicidad inaceptable.
Tome STIVARGA a la misma hora cada día. Trague el comprimido entero con agua después de una comida baja en grasas que contenga menos de 600 calorías y menos del 30% de grasas [ver Farmacología Clínica (12.3)]. No tome dos dosis de STIVARGA en el mismo día para compensar una dosis olvidada del día anterior.
2.2 Modificaciones de dosis
Si se requieren modificaciones de dosis, reduzca la dosis en incrementos de 40 mg (un comprimido); la dosis diaria más baja recomendada de STIVARGA es de 80 mg al día.
Interrumpa STIVARGA en los siguientes casos:
- •
- Reacción cutánea mano-pie (HFSR) [síndrome de eritrodisestesia palmo-plantar (PPES)] de Grado 2 que es recurrente o no mejora dentro de los 7 días a pesar de la reducción de la dosis; interrumpa el tratamiento durante un mínimo de 7 días para HFSR de Grado 3
- •
- Hipertensión sintomática de Grado 2
- •
- Cualquier reacción adversa de Grado 3 o 4
- •
- Empeoramiento de la infección de cualquier grado
Reduzca la dosis de STIVARGA a 120 mg:
- •
- Para la primera aparición de HFSR de Grado 2 de cualquier duración
- •
- Después de la recuperación de cualquier reacción adversa de Grado 3 o 4, excepto infección
- •
- Para elevación de aspartato aminotransferasa (AST)/alanina aminotransferasa (ALT) de Grado 3, solo reanude si el beneficio potencial supera el riesgo de hepatotoxicidad
Reduzca la dosis de STIVARGA a 80 mg:
- •
- Para la reaparición de HFSR de Grado 2 con la dosis de 120 mg
- •
- Después de la recuperación de cualquier reacción adversa de Grado 3 o 4 con la dosis de 120 mg (excepto hepatotoxicidad o infección)
Suspenda STIVARGA permanentemente en los siguientes casos:
- •
- Incapacidad para tolerar la dosis de 80 mg
- •
- Cualquier aparición de AST o ALT más de 20 veces el límite superior de lo normal (ULN)
- •
- Cualquier aparición de AST o ALT más de 3 veces el ULN con bilirrubina concurrente más de 2 veces el ULN
- •
- Reaparición de AST o ALT más de 5 veces el ULN a pesar de la reducción de dosis a 120 mg
- •
- Para cualquier reacción adversa de Grado 4; solo reanude si el beneficio potencial supera los riesgos
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
STIVARGA es una tableta recubierta con película de 40 mg, de color rosa claro, de forma ovalada, con la inscripción ‘BAYER’ en un lado y ’40’ en el otro lado.
4 CONTRAINDICACIONES
Ninguna.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Hepatotoxicidad
En ensayos clínicos ocurrió lesión hepática grave inducida por fármacos con desenlace fatal en pacientes tratados con STIVARGA. En la mayoría de los casos, la disfunción hepática ocurrió dentro de los 2 primeros meses de terapia y se caracterizó por un patrón hepatocelular de lesión.
En el estudio CORRECT, se produjo insuficiencia hepática fatal en el 1.6% de los pacientes en el brazo de regorafenib y en el 0.4% de los pacientes en el brazo de placebo. En el estudio GRID, se produjo insuficiencia hepática fatal en el 0.8% de los pacientes en el brazo de regorafenib. En el estudio RESORCE, no hubo aumento en la incidencia de insuficiencia hepática fatal en comparación con el placebo [ver Reacciones Adversas (6.1)].
Obtenga pruebas de función hepática (ALT, AST y bilirrubina) antes de iniciar STIVARGA y controle al menos cada dos semanas durante los primeros 2 meses de tratamiento. A partir de entonces, controle mensualmente o con mayor frecuencia según esté clínicamente indicado. Controle las pruebas de función hepática semanalmente en pacientes que experimentan elevación de las pruebas de función hepática hasta que mejoren a menos de 3 veces el LSN o el valor inicial.
Suspenda temporalmente y luego reduzca o suspenda permanentemente STIVARGA según la gravedad y la persistencia de la hepatotoxicidad manifestada por la elevación de las pruebas de función hepática o necrosis hepatocelular [ver Dosis y Administración (2.2) y Uso en Poblaciones Específicas (8.6)].
5.2 Infecciones
STIVARGA causó un mayor riesgo de infecciones. La incidencia general de infección (Grados 1-5) fue más alta (32% vs. 17%) en 1142 pacientes tratados con STIVARGA en comparación con el brazo de control en ensayos aleatorios controlados con placebo. La incidencia de infecciones de grado 3 o superior en pacientes tratados con STIVARGA fue del 9%. Las infecciones más comunes fueron infecciones del tracto urinario (5.7%), nasofaringitis (4.0%), infecciones fúngicas mucocutáneas y sistémicas (3.3%) y neumonía (2.6%). Los desenlaces fatales causados por infección ocurrieron con mayor frecuencia en los pacientes tratados con STIVARGA (1.0%) en comparación con los pacientes que recibieron placebo (0.3%); las infecciones fatales más comunes fueron respiratorias (0.6% en los pacientes tratados con STIVARGA frente al 0.2% en los pacientes que recibieron placebo).
Suspenda STIVARGA en caso de infecciones de Grado 3 o 4, o empeoramiento de la infección de cualquier grado. Reanude STIVARGA a la misma dosis después de la resolución de la infección [ver Dosis y Administración (2.2)].
5.3 Hemorragia
STIVARGA causó una mayor incidencia de hemorragia. La incidencia general (Grados 1-5) fue del 18.2% en 1142 pacientes tratados con STIVARGA y del 9.5% en pacientes que recibieron placebo en ensayos aleatorios controlados con placebo. La incidencia de hemorragia de grado 3 o superior en pacientes tratados con STIVARGA fue del 3.0%. La incidencia de eventos hemorrágicos fatales fue del 0.7%, involucrando el sistema nervioso central o los tractos respiratorio, gastrointestinal o genitourinario.
Suspenda permanentemente STIVARGA en pacientes con hemorragia grave o potencialmente mortal. Controle los niveles de INR con mayor frecuencia en los pacientes que reciben warfarina [ver Farmacología Clínica (12.3)].
5.4 Perforación Gastrointestinal o Fístula
La perforación gastrointestinal ocurrió en el 0.6% de 4518 pacientes tratados con STIVARGA en todos los ensayos clínicos de STIVARGA administrado como agente único; esto incluyó ocho eventos fatales.
La fístula gastrointestinal ocurrió en el 0.8% de los pacientes tratados con STIVARGA y en el 0.2% de los pacientes en el brazo de placebo en todos los ensayos aleatorios controlados con placebo. Suspenda permanentemente STIVARGA en pacientes que desarrollen perforación gastrointestinal o fístula.
5.5 Toxicidad Dermatológica
En ensayos aleatorios controlados con placebo, las reacciones adversas cutáneas ocurrieron en el 71.9% de los pacientes en el brazo de regorafenib y en el 25.5% de los pacientes en el brazo de placebo, incluida la reacción cutánea mano-pie (HFSR, por sus siglas en inglés) también conocida como síndrome de eritrodisestesia palmo-plantar (PPES, por sus siglas en inglés), y erupción cutánea grave que requiere modificación de la dosis.
En los ensayos aleatorios controlados con placebo, la incidencia general de HFSR fue mayor en los 1142 pacientes tratados con STIVARGA (53%) que en los pacientes tratados con placebo (8%). La mayoría de los casos de HFSR en pacientes tratados con STIVARGA aparecieron durante el primer ciclo de tratamiento. Las incidencias de HFSR Grado 3 (16% versus <1%), erupción cutánea Grado 3 (3% versus <1%), reacciones adversas graves de eritema multiforme (<0.1% vs. 0%) y síndrome de Stevens-Johnson (<0.1% vs. 0%) también fueron más altas en los pacientes tratados con STIVARGA [ver Reacciones Adversas (6.1)]. En todos los ensayos, se observó una mayor incidencia de HFSR en los pacientes asiáticos tratados con STIVARGA (todos los grados: 72%; Grado 3: 18%) [ver Uso en Poblaciones Específicas (8.8)].
La necrólisis epidérmica tóxica ocurrió en el 0.02% de los 4518 pacientes tratados con STIVARGA en todos los ensayos clínicos de STIVARGA administrado como agente único.
Suspenda, reduzca la dosis o suspenda permanentemente STIVARGA según la gravedad y la persistencia de la toxicidad dermatológica [ver Dosis y Administración (2.2)]. Instituya medidas de apoyo para el alivio sintomático.
5.6 Hipertensión
En ensayos aleatorios controlados con placebo, se produjo crisis hipertensiva en el 0.2% de los pacientes en los brazos de regorafenib y en ninguno de los pacientes en los brazos de placebo. STIVARGA causó una mayor incidencia de hipertensión (30% versus 8% en CORRECT, 59% versus 27% en GRID y 31% versus 6% en RESORCE) [ver Reacciones Adversas (6.1)]. El inicio de la hipertensión ocurrió durante el primer ciclo de tratamiento en la mayoría de los pacientes que desarrollaron hipertensión (67% en ensayos aleatorios controlados con placebo).
No inicie STIVARGA a menos que la presión arterial esté controlada adecuadamente. Controle la presión arterial semanalmente durante las primeras 6 semanas de tratamiento y luego en cada ciclo, o con mayor frecuencia, según esté clínicamente indicado. Suspenda temporal o permanentemente STIVARGA en caso de hipertensión grave o no controlada [ver Dosis y Administración (2.2)].
5.7 Isquemia e infarto cardíaco
STIVARGA aumentó la incidencia de isquemia e infarto de miocardio (0,9 % frente a 0,2 %) en ensayos aleatorizados controlados con placebo [ver Reacciones adversas (6.1)]. Suspenda STIVARGA en pacientes que desarrollen isquemia cardíaca nueva o de inicio agudo o infarto. Reanude STIVARGA solo después de la resolución de los eventos isquémicos cardíacos agudos, si los beneficios potenciales superan los riesgos de isquemia cardíaca adicional.
5.8 Síndrome de leucoencefalopatía posterior reversible
El síndrome de leucoencefalopatía posterior reversible (RPLS), un síndrome de edema vasogénico subcortical diagnosticado por hallazgos característicos en la resonancia magnética, ocurrió en uno de cada 4800 pacientes tratados con STIVARGA en todos los ensayos clínicos. Realice una evaluación de RPLS en cualquier paciente que presente convulsiones, dolor de cabeza intenso, alteraciones visuales, confusión o alteración de la función mental. Discontinúe STIVARGA en pacientes que desarrollen RPLS.
5.9 Riesgo de deterioro de la cicatrización de heridas
Pueden ocurrir complicaciones de cicatrización de heridas deteriorada en pacientes que reciben medicamentos que inhiben la vía de señalización VEGF. Por lo tanto, STIVARGA tiene el potencial de afectar negativamente la cicatrización de heridas.
Suspenda STIVARGA durante al menos 2 semanas antes de la cirugía electiva. No administrar durante al menos 2 semanas después de una cirugía mayor y hasta una cicatrización adecuada de la herida. No se ha establecido la seguridad de reanudar STIVARGA después de la resolución de las complicaciones de la cicatrización de heridas.
5.10 Toxicidad embriofetal
Basado en estudios en animales y su mecanismo de acción, STIVARGA puede causar daño fetal cuando se administra a una mujer embarazada. No hay datos disponibles sobre el uso de STIVARGA en mujeres embarazadas. Regorafenib fue embrioletal y teratogénico en ratas y conejos a exposiciones más bajas que las exposiciones humanas a la dosis recomendada, con una mayor incidencia de malformaciones cardiovasculares, genitourinarias y esqueléticas. Informe a las mujeres embarazadas del riesgo potencial para el feto.
Aconseje a las mujeres en edad reproductiva que usen anticonceptivos efectivos durante el tratamiento con STIVARGA y durante 2 meses después de la dosis final. Aconseje a los hombres con parejas femeninas en edad reproductiva que usen anticonceptivos efectivos durante el tratamiento con STIVARGA y durante 2 meses después de la dosis final [ver Uso en poblaciones específicas (8.1), (8.3)].
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas graves se discuten en otras secciones del etiquetado:
- •
- Hepatotoxicidad [ver Advertencias y precauciones (5.1)]
- •
- Infecciones [ver Advertencias y precauciones (5.2)]
- •
- Hemorragia [ver Advertencias y precauciones (5.3)]
- •
- Perforación gastrointestinal o fístula [ver Advertencias y precauciones (5.4)]
- •
- Toxicidad dermatológica [ver Advertencias y precauciones (5.5)]
- •
- Hipertensión [ver Advertencias y precauciones (5.6)]
- •
- Isquemia cardíaca e infarto [ver Advertencias y precauciones (5.7)]
- •
- Síndrome de leucoencefalopatía posterior reversible (RPLS) [ver Advertencias y precauciones (5.8)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no pueden compararse directamente con las tasas de los ensayos clínicos de otro fármaco y pueden no reflejar la tasa observada en la práctica.
Los datos descritos en la sección de ADVERTENCIAS Y PRECAUCIONES reflejan la exposición a STIVARGA en más de 4800 pacientes que fueron inscritos en cuatro ensayos aleatorizados, controlados con placebo (n=1142), un programa de acceso ampliado (CONSIGN, n=2864) o ensayos clínicos de un solo brazo (agente único o en combinación con otros agentes). Hubo 4518 pacientes que recibieron STIVARGA como agente único; la distribución de neoplasias subyacentes fue 80% CCR, 4% GIST, 10% CHC, 6% otros tumores sólidos; y 74% eran caucásicos, 11% asiáticos y 15% de raza desconocida. Entre estos 4518 pacientes, el 83% recibió STIVARGA durante al menos 21 días y el 20% recibió STIVARGA durante 6 meses o más.
En ensayos aleatorizados controlados con placebo (CORRECT, GRID, RESORCE y CONCUR), las reacciones adversas al fármaco observadas con mayor frecuencia (≥20%) en pacientes que reciben STIVARGA son dolor (incluyendo dolor gastrointestinal y abdominal), HFSR, astenia/fatiga, diarrea, disminución del apetito/ingesta de alimentos, hipertensión, infección, disfonía, hiperbilirrubinemia, fiebre, mucositis, pérdida de peso, erupción cutánea y náuseas.
Cáncer colorrectal
Los datos de seguridad que se describen a continuación, excepto donde se indique, se derivan de un ensayo aleatorizado (2:1), doble ciego, controlado con placebo (CORRECT) en el que 500 pacientes (mediana de edad 61 años; 61% hombres) con cáncer colorrectal metastásico (CCR) previamente tratado recibieron STIVARGA como agente único a la dosis de 160 mg diarios durante las primeras 3 semanas de cada ciclo de tratamiento de 4 semanas y 253 pacientes (mediana de edad 61 años; 60% hombres) recibieron placebo. La duración media de la terapia fue de 1,7 meses (rango 2 días, 10,8 meses) para los pacientes que recibieron STIVARGA. Debido a las reacciones adversas, el 61% de los pacientes que recibieron STIVARGA requirieron una interrupción de la dosis y el 38% de los pacientes tuvieron su dosis reducida. Las reacciones adversas que resultaron en la interrupción del tratamiento ocurrieron en el 8,2% de los pacientes tratados con STIVARGA en comparación con el 1,2% de los pacientes que recibieron placebo. La reacción cutánea mano-pie (HFSR) y la erupción cutánea fueron las razones más comunes para la interrupción permanente de STIVARGA.
La Tabla 1 proporciona la incidencia de reacciones adversas (≥10%) en pacientes en CORRECT.
|
Reacciones adversas |
STIVARGA (N=500) |
Placebo (N=253) |
||
|---|---|---|---|---|
| Grado | Grado | |||
| Todos % |
≥ 3 % |
Todos % |
≥ 3 % |
|
|
Trastornos generales y alteraciones en el lugar de administración Astenia/fatiga Dolor Fiebre |
64 59 28 |
15 9 2 |
46 48 15 |
9 7 0 |
|
Trastornos del metabolismo y de la nutrición Disminución del apetito y de la ingesta de alimentos |
47 |
5 |
28 |
4 |
|
Trastornos de la piel y del tejido subcutáneo HFSR/PPES Erupción cutánea b |
45 26 |
17 6 |
7 4 |
0 <1 |
|
Trastornos gastrointestinales Diarrea Mucositis |
43 33 |
8 4 |
17 5 |
2 0 |
|
Investigaciones Pérdida de peso |
32 |
<1 |
10 |
0 |
|
Infecciones e infestaciones Infección c |
31 |
9 |
17 |
6 |
|
Trastornos vasculares Hipertensión Hemorragia c |
30 21 |
8 2 |
8 8 |
<1 <1 |
|
Trastornos respiratorios, torácicos y mediastínicos Disfonía |
30 |
0 |
6 |
0 |
|
Trastornos del sistema nervioso Dolor de cabeza |
10 |
<1 |
7 |
0 |
- a Reacciones adversas clasificadas de acuerdo a los Criterios Comunes de Toxicidad para Eventos Adversos del National Cancer Institute versión 3.0 (NCI CTCAE v3.0).
- bEl término erupción representa reportes de eventos de erupción medicamentosa, erupción, erupción eritematosa, erupción generalizada, erupción macular, erupción maculo-papular, erupción papular y erupción prurítica.
cSe observaron desenlaces fatales.
La Tabla 2 proporciona las anormalidades de laboratorio observadas en CORRECT.
| Parámetro de laboratorio | STIVARGA (N=500 a) |
Placebo (N=253 a) |
||||
|---|---|---|---|---|---|---|
| Grado b | Grado b | |||||
| Todo % |
3 % |
4 % |
Todo % |
3 % |
4 % |
|
|
Trastornos sanguíneos y del sistema linfático |
||||||
|
Anemia |
79 |
5 |
1 |
66 |
3 |
0 |
|
Trombocitopenia |
41 |
2 |
<1 |
17 |
<1 |
0 |
|
Neutropenia |
3 |
1 |
0 |
0 |
0 |
0 |
|
54 |
9 |
0 |
35 |
4 |
<1 |
|
Trastornos del metabolismo y de la nutrición |
||||||
|
Hipocalcemia |
59 |
1 |
<1 |
18 |
1 |
0 |
|
Hipopotasemia |
26 |
4 |
0 |
8 |
<1 |
0 |
|
Hiponatremia |
30 |
7 |
1 |
22 |
4 |
0 |
|
Hipofosfatemia |
57 |
31 |
1 |
11 |
4 |
0 |
|
Trastornos hepatobiliares |
||||||
|
Hiperbilirrubinemia |
45 |
10 |
3 |
17 |
5 |
3 |
|
Aumento de AST |
65 |
5 |
1 |
46 |
4 |
1 |
|
Aumento de ALT |
45 |
5 |
1 |
30 |
3 |
<1 |
|
Trastornos renales y urinarios |
||||||
|
Proteinuriac |
84 |
2 |
0 |
61 |
1 |
0 |
|
Investigaciones |
||||||
|
Aumento del INRd |
24 |
4 |
N/A |
17 |
2 |
N/A |
|
Aumento de Lipasa |
46 |
9 |
2 |
19 |
3 |
2 |
|
Aumento de Amilasa |
26 |
2 |
<1 |
17 |
2 |
<1 |
a % basado en el número de pacientes con muestras post-basal que pueden ser menos de 500 (regorafenib) o 253 (placebo).
b NCI CTCAE v3.0.
cBasado en datos de la relación proteína-creatinina en orina.
dInternational normalized ratio: No se denota Grado 4 en NCI CTCAE, v3.0.
Tumores del Estroma Gastrointestinal
Los datos de seguridad descritos a continuación se derivan de un ensayo aleatorizado (2:1), doble ciego, controlado con placebo (GRID) en el que 132 pacientes (mediana de edad 60 años; 64% hombres) con GIST previamente tratado recibieron STIVARGA como agente único a una dosis de 160 mg diarios durante las primeras 3 semanas de cada ciclo de tratamiento de 4 semanas y 66 pacientes (mediana de edad 61 años; 64% hombres) recibieron placebo. La duración media de la terapia fue de 5,7 meses (rango 1 día, 11,7 meses) para los pacientes que recibieron STIVARGA. Se requirieron interrupciones de dosis por eventos adversos en el 58% de los pacientes que recibieron STIVARGA y el 50% de los pacientes tuvieron su dosis reducida. Las reacciones adversas que resultaron en la interrupción del tratamiento se informaron en el 2,3% de los pacientes tratados con STIVARGA en comparación con el 1,5% de los pacientes que recibieron placebo.
La Tabla 3 proporciona la incidencia de reacciones adversas (≥10%) en pacientes en GRID.
| Reacciones Adversas | STIVARGA (N=132) |
Placebo (N=66) |
||||||
|---|---|---|---|---|---|---|---|---|
| Grado | Grado | |||||||
| Todos % |
≥ 3 % |
Todos % |
≥ 3 % |
|||||
|
Trastornos de la piel y del tejido subcutáneo HFSR/PPE Erupciónb Alopecia |
67 30 24 |
22 7 2 |
12 3 2 |
2 0 0 |
||||
|
Trastornos generales y afecciones en el lugar de administración Astenia/Fatiga Fiebre |
52 21 |
4 0 |
39 11 |
2 2 |
||||
|
Trastornos vasculares Hipertensión Hemorragia |
59 11 |
28 4 |
27 3 |
5 0 |
||||
|
Trastornos gastrointestinales Dolor Diarrea Mucositis Náuseas Vómitos |
60 47 40 20 17 |
8 8 2 2 <1 |
55 9 8 12 8 |
Trastornos respiratorios, torácicos y mediastínicos Disfonía |
39 |
0 |
9 |
0 |
|
Infecciones e infestaciones Infección c |
32 |
5 |
5 |
0 |
||||
|
Trastornos del metabolismo y de la nutrición Disminución del apetito e ingesta de alimentos Hipotiroidismo d |
31 18 |
<1 0 |
21 6 |
3 0 |
||||
|
Trastornos del sistema nervioso Dolor de cabeza |
16 |
0 |
9 |
0 |
||||
|
Investigaciones Pérdida de peso |
14 |
0 |
8 |
0 |
||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo Espasmos musculares |
14 |
0 |
3 |
0 |
||||
a Reacciones adversas clasificadas de acuerdo con NCI CTCAE v4.0.
- bEl término erupción cutánea representa reportes de eventos de erupción cutánea, erupción eritematosa, erupción macular, erupción maculopapular, erupción papular y erupción prurítica.
- cSe observaron desenlaces fatales.
dIncidencia de hipotiroidismo basada en un subconjunto de pacientes con TSH normal y sin suplementación tiroidea al inicio del estudio.
La Tabla 4 proporciona anormalidades de laboratorio observadas en GRID.
| Parámetro de laboratorio | STIVARGA (N=132a) |
Placebo (N=66 a) |
||||
|---|---|---|---|---|---|---|
| Grado b | Grado b | |||||
| Todos % |
3 % |
4 % |
Todos % |
3 % |
4 % |
|
|
Trastornos de la sangre y del sistema linfático Trombocitopenia Neutropenia Linfopenia |
13 16 30 |
1 2 8 |
0 1 0 |
2 12 24 |
0 3 3 |
2 0 0 |
|
Trastornos del metabolismo y de la nutrición Hipocalcemia Hipokalemia Hipofosfatemia |
17 21 55 |
2 3 20 |
0 0 2 |
5 3 3 |
0 0 2 |
0 0 0 |
|
Trastornos hepatobiliares Hiperbilirrubinemia AST aumentada ALT aumentada |
33 58 39 |
3 3 4 |
1 1 1 |
12 47 39 |
2 3 2 |
0 0 0 |
|
Trastornos renales y urinarios Proteinuria c |
59 |
3 |
– d |
53 |
3 |
– d |
|
Investigaciones Lipasa aumentada |
14 |
0 |
1 |
5 |
0 |
0 |
- a Porcentaje basado en el número de pacientes con muestras post-basales que pueden ser menos de 132 (regorafenib) o 66 (placebo).
b NCI CTCAE v4.0.
c Basado en datos de relación proteína/creatinina en orina.
d No se denota Grado 4 en NCI CTCAE v4.0.
Carcinoma Hepatocelular
Los datos de seguridad que se describen a continuación se derivan de un ensayo aleatorizado (2:1), doble ciego, controlado con placebo (RESORCE) en el que pacientes con CHC previamente tratado recibieron STIVARGA (n=374) 160 mg por vía oral los días 1-21 de cada ciclo de tratamiento de 4 semanas o placebo (n=193). La mediana de edad fue de 63 años, el 88% eran hombres, el 98% tenía cirrosis Child-Pugh A, el 66% tenía un estado funcional ECOG (EF) de 0 y el 34% tenía un EF de 1. La duración media del tratamiento fue de 3,5 meses (rango de 1 día a 29,4 meses) para los pacientes que recibieron STIVARGA. De los pacientes que recibieron STIVARGA, el 33% estuvo expuesto a STIVARGA durante ≥6 meses y el 14% estuvo expuesto a STIVARGA durante ≥12 meses. Se requirieron interrupciones de la dosis por eventos adversos en el 58,3% de los pacientes que recibieron STIVARGA y el 48% de los pacientes tuvieron su dosis reducida. Las reacciones adversas más comunes que requirieron modificación de la dosis (interrupción o reducción de la dosis) fueron HFSR/PPES (20,6%), aumento de la bilirrubina en sangre (5,9%), fatiga (5,1%) y diarrea (5,3%). Se notificaron reacciones adversas que dieron lugar a la interrupción del tratamiento en el 10,4% de los pacientes tratados con STIVARGA en comparación con el 3,6% de los pacientes que recibieron placebo; las reacciones adversas más frecuentes que requirieron la interrupción de STIVARGA fueron HFSR/PPES (1,9%) y aumento de AST (1,6%).
La Tabla 5 proporciona la incidencia de reacciones adversas (≥10%) en pacientes en RESORCE.
| Reacciones Adversas | STIVARGA (N=374) |
Placebo (N=193) |
||
|---|---|---|---|---|
| Grado | Grado | |||
| Todas % |
≥ 3 % |
Todas % |
≥ 3 % |
|
|
Trastornos de la piel y del tejido subcutáneo HFSR/PPE |
51 |
12 |
7 |
<1 |
|
Trastornos generales y afecciones en el lugar de administración Dolor Astenia/Fatiga Fiebre |
55 42 20 |
9 10 0 |
44 33 7 |
8 5 0 |
|
Trastornos vasculares Hipertensión Hemorragiab |
31 18 |
15 5 |
6 16 |
5 8 |
|
Trastornos gastrointestinales Diarrea Náuseas Vómitos Mucositis |
41 17 13 13 |
3 <1 <1 1 |
15 13 7 2 |
0 0 <1 ≤1 |
|
Trastornos respiratorios, torácicos y mediastínicos Disfonía |
18 |
0 |
2 |
0 |
|
Infecciones e infestaciones Infección b |
31 |
8 |
18 |
6 |
|
Trastornos del metabolismo y de la nutrición Disminución del apetito y de la ingesta de alimentos |
31 |
3 |
15 |
2 |
|
Investigaciones Pérdida de peso |
13 |
2 |
4 |
0 |
|
Espasmos musculares |
10 |
0 |
2 |
0 |
a Reacciones adversas clasificadas según NCI CTCAE v4.0.
- bSe observaron desenlaces fatales.
Otras reacciones adversas clínicamente significativas observadas en menos del 10% de los pacientes tratados con STIVARGA fueron: alopecia (7%), hipotiroidismo (6,4%), pancreatitis (1,6%), erupción exfoliativa (1,3%), temblor (1,3%), eritema multiforme (0,8%), isquemia miocárdica (0,8%), fístula gastrointestinal (0,3%) e infarto de miocardio (0,3%).
La Tabla 6 proporciona las anomalías de laboratorio observadas en RESORCE.
| Parámetro de laboratorio | STIVARGA (N=374a) |
Placebo (N=193 a) |
||||
|---|---|---|---|---|---|---|
| Grado b | Grado b | |||||
| Todos % |
3 % |
4 % |
Todos % |
3 % |
4 % |
|
|
Trastornos de la sangre y del sistema linfático Trombocitopenia Neutropenia Linfopenia |
63 14 68 |
5 3 16 |
<1 0 2 |
50 15 59 |
0 <1 11 |
0 <1 <1 |
|
Trastornos del metabolismo y de la nutrición Hipocalcemia Hipopotasemia Hipofosfatemia |
23 31 70 |
<1 4 32 |
0 <1 2 |
10 9 31 |
0 2 7 |
0 0 0 |
|
Trastornos hepatobiliares Hiperbilirrubinemia Aumento de AST Aumento de ALT |
78 93 70 |
13 16 6 6 REACCIONES ADVERSAS |
3 2 <1 |
55 84 59 |
11 17 5 |
5 3 0 |
|
Trastornos renales y urinarios Proteinuria c |
51 |
17 |
– d |
37 |
3 |
– d |
|
Investigaciones Aumento del INR Aumento de Lipasa Aumento de Amilasa |
44 41 23 |
<1 11 3 |
– d 3 <1 |
35 27 19 |
2 8 2 |
– d 1 <1 |
- a Porcentaje basado en el número de pacientes con muestras post-basal que puede ser menor que 374 (regorafenib) o 193 (placebo).
b NCI CTCAE v4.0.
c Basado en datos de tira reactiva.
d No se denota Grado 4 en NCI CTCAE v4.0.
6.2 Experiencia postcomercialización
Se ha identificado la siguiente reacción adversa durante el uso posaprobación de STIVARGA. Debido a que estas reacciones se reportan voluntariamente a partir de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco:
- •
- reacción de hipersensibilidad
- •
- síndrome nefrótico
- •
- insuficiencia cardíaca
- •
- aneurismas arteriales (incluida la aorta), disecciones y rotura
7 INTERACCIONES MEDICAMENTOSAS
7.1 Efecto de los Inductores Potentes de CYP3A4 sobre Regorafenib
La administración conjunta de un inductor potente de CYP3A4 con STIVARGA disminuyó las concentraciones plasmáticas de regorafenib, aumentó las concentraciones plasmáticas del metabolito activo M-5 y no produjo cambios en las concentraciones plasmáticas del metabolito activo M-2 [ver Farmacología Clínica (12.3)], y puede conducir a una disminución de la eficacia. Evite el uso concomitante de STIVARGA con inductores potentes de CYP3A4 (por ejemplo, rifampicina, fenitoína, carbamazepina, fenobarbital y hierba de San Juan).
7.2 Efecto de los Inhibidores Potentes de CYP3A4 sobre Regorafenib
La administración conjunta de un inhibidor potente de CYP3A4 con STIVARGA aumentó las concentraciones plasmáticas de regorafenib y disminuyó las concentraciones plasmáticas de los metabolitos activos M-2 y M-5 [ver Farmacología Clínica (12.3)], y puede conducir a una mayor toxicidad. Evite el uso concomitante de STIVARGA con inhibidores potentes de CYP3A4 (por ejemplo, claritromicina, jugo de toronja, itraconazol, ketoconazol, nefazodona, posaconazol, telitromicina y voriconazol).
7.3 Efecto de Regorafenib sobre los Sustratos de la Proteína de Resistencia al Cáncer de Mama (BCRP)
La administración conjunta de STIVARGA con un sustrato de BCRP aumentó las concentraciones plasmáticas del sustrato de BCRP [ver Farmacología Clínica (12.3)]. Monitoree de cerca a los pacientes para detectar signos y síntomas de toxicidad relacionada con la exposición al sustrato de BCRP (por ejemplo, metotrexato, fluvastatina, atorvastatina). Consulte la información del producto del sustrato de BCRP concomitante cuando considere la administración de dichos productos junto con STIVARGA.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de riesgos
Con base en estudios en animales y su mecanismo de acción, STIVARGA puede causar daño fetal cuando se administra a una mujer embarazada. No hay datos disponibles sobre el uso de STIVARGA en mujeres embarazadas. La administración de regorafenib fue embrioletal y teratogénica en ratas y conejos a exposiciones menores que las exposiciones humanas a la dosis recomendada, con mayores incidencias de malformaciones cardiovasculares, genitourinarias y esqueléticas [ver Datos]. Informe a las mujeres embarazadas del peligro potencial para el feto.
Se desconoce el riesgo de referencia estimado de defectos congénitos importantes y aborto espontáneo para la población indicada. En la población general de los EE. UU., el riesgo de referencia estimado de defectos congénitos importantes y aborto espontáneo en embarazos clínicamente reconocidos es del 2 al 4 % y del 15 al 20 %, respectivamente.
Datos
Datos en animales
En estudios de desarrollo embriofetal, se observó una pérdida total del embarazo (reabsorción del 100 % de la camada) en ratas a dosis tan bajas como 1 mg/kg (aproximadamente el 6 % de la dosis humana recomendada, según el área de superficie corporal) y en conejos a dosis tan bajas como 1,6 mg/kg (aproximadamente el 25 % de la exposición humana a la dosis clínicamente recomendada medida por AUC).
En un estudio de distribución de dosis única en ratas embarazadas, hubo una mayor penetración de regorafenib a través de la barrera hematoencefálica en los fetos en comparación con las madres. La administración diaria de regorafenib a ratas embarazadas durante la organogénesis resultó en hallazgos fetales de osificación retardada a dosis > 0,8 mg/kg (aproximadamente el 5 % de la dosis humana recomendada según el área de superficie corporal) y aumentos dependientes de la dosis en malformaciones esqueléticas, incluido paladar hendido y fontanela agrandada a dosis ≥ 1 mg/kg (aproximadamente el 10 % de la exposición clínica basada en AUC). A dosis ≥ 1,6 mg/kg (aproximadamente el 11 % de la dosis humana recomendada según el área de superficie corporal), hubo aumentos dependientes de la dosis en la incidencia de malformaciones cardiovasculares, anomalías externas, hernia diafragmática y dilatación de la pelvis renal.
En conejas embarazadas a las que se les administró regorafenib diariamente durante la organogénesis, se observaron defectos del tabique ventricular a la dosis más baja probada de 0,4 mg/kg (aproximadamente el 7 % del AUC en pacientes a la dosis recomendada). A dosis de ≥ 0,8 mg/kg (aproximadamente el 15 % de la exposición humana a la dosis humana recomendada según el AUC), la administración de regorafenib resultó en aumentos dependientes de la dosis en la incidencia de malformaciones cardiovasculares y anomalías esqueléticas adicionales, así como efectos adversos significativos en el sistema urinario, incluyendo riñón/uréter faltante, riñón pequeño, deformado y mal posicionado, e hidronefrosis. La proporción de fetos viables que eran machos disminuyó al aumentar la dosis en dos estudios de toxicidad embriofetal en conejos.
8.2 Lactancia
Resumen de riesgos
No hay datos sobre la presencia de regorafenib o sus metabolitos en la leche materna, los efectos de regorafenib en el lactante amamantado o sobre la producción de leche. En ratas, regorafenib y sus metabolitos se excretan en la leche. Debido al potencial de reacciones adversas graves en lactantes amamantados por STIVARGA, no amamante durante el tratamiento con STIVARGA y durante 2 semanas después de la dosis final.
8.3 Mujeres y hombres con capacidad reproductiva
Anticoncepción
Mujeres
Use anticonceptivos eficaces durante el tratamiento y durante 2 meses después de completar la terapia.
Hombres
Aconseje a los pacientes masculinos con parejas femeninas con potencial reproductivo que usen anticonceptivos eficaces durante el tratamiento y durante 2 meses después de la dosis final de STIVARGA [ver Toxicología no clínica (13.1)].
Infertilidad
No hay datos sobre el efecto de STIVARGA sobre la fertilidad humana. Los resultados de estudios en animales indican que regorafenib puede perjudicar la fertilidad masculina y femenina [ver Toxicología no clínica (13.1)].
8.4 Uso pediátrico
No se ha establecido la seguridad y eficacia de STIVARGA en pacientes pediátricos menores de 18 años.
Datos en animales
En estudios de dosis repetidas de 28 días en ratas, hubo hallazgos dependientes de la dosis de alteración de la dentina y angiectasia. Estos hallazgos ocurrieron con dosis de regorafenib tan bajas como 4 mg/kg (aproximadamente el 25 % del AUC en humanos a la dosis recomendada). En estudios de dosis repetidas de 13 semanas en perros, hubo hallazgos similares de alteración de la dentina con dosis tan bajas como 20 mg/kg (aproximadamente el 43 % del AUC en humanos a la dosis recomendada). La administración de regorafenib en estos animales también condujo a un crecimiento persistente y engrosamiento de la placa de crecimiento epifisario femoral.
8.5 Uso geriátrico
De los 1142 pacientes tratados con STIVARGA inscritos en ensayos aleatorizados y controlados con placebo, el 40 % tenía 65 años o más, mientras que el 10 % tenía 75 años o más. No se observaron diferencias generales en la eficacia entre estos pacientes y los pacientes más jóvenes. Hubo una mayor incidencia de hipertensión de Grado 3 (18 % frente a 9 %) en los ensayos controlados con placebo entre los pacientes tratados con STIVARGA de 65 años o más en comparación con los pacientes más jóvenes. Además, se ha informado un evento de hipertensión de Grado 4 en el grupo de edad de 65 años o más y ninguno en el grupo de edad más joven.
8.6 Insuficiencia hepática
No se recomienda ajustar la dosis en pacientes con insuficiencia hepática leve (bilirrubina total ≤LSN y AST >LSN, o bilirrubina total >LSN a ≤1,5 veces el LSN) o moderada (bilirrubina total >1,5 a ≤3 veces el LSN y cualquier AST), [ver Farmacología clínica (12.3)]. Monitoree de cerca a los pacientes con insuficiencia hepática para detectar reacciones adversas [ver Advertencias y precauciones (5.1)].
No se recomienda el uso de STIVARGA en pacientes con insuficiencia hepática grave (bilirrubina total >3 veces el LSN) ya que STIVARGA no se ha estudiado en esta población.
8.7 Insuficiencia renal
No se recomienda ajustar la dosis para pacientes con insuficiencia renal. La farmacocinética de regorafenib no se ha estudiado en pacientes que están en diálisis y no hay una dosis recomendada para esta población de pacientes [ver Farmacología clínica (12.3)].
8.8 Raza
Según los datos agrupados de tres ensayos controlados con placebo (CORRECT, GRID y CONCUR), se produjo una mayor incidencia de HFSR y anomalías en las pruebas de función hepática en pacientes asiáticos tratados con STIVARGA en comparación con los blancos [ver Advertencias y precauciones (5.1, 5.5)]. No es necesario ajustar la dosis inicial según la raza.
10 SOBREDOSIS
La dosis más alta de STIVARGA estudiada clínicamente es 220 mg por día. Las reacciones adversas al medicamento observadas con más frecuencia a esta dosis fueron eventos dermatológicos, disfonía, diarrea, inflamación de la mucosa, sequedad de boca, disminución del apetito, hipertensión y fatiga. No se conoce ningún antídoto para la sobredosis de STIVARGA. En caso de sospecha de sobredosis, interrumpa STIVARGA, instituya atención de apoyo y observe hasta la estabilización clínica.
11 DESCRIPCIÓN
STIVARGA (regorafenib) es un inhibidor de multicinasas con el nombre químico 4-[4-({[4-cloro-3-(trifluorometil) fenil] carbamoil} amino)-3-fluorofenoxi]-N-metilpiridina-2-carboxamida monohidrato. Regorafenib tiene la siguiente fórmula estructural:
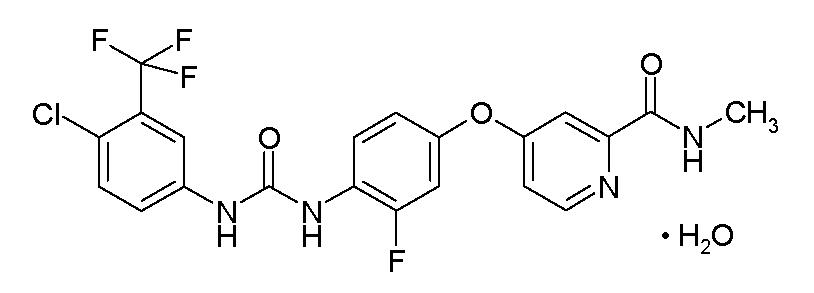
Regorafenib es un monohidrato y tiene una fórmula molecular C21H15ClF4N4O3 • H2O y un peso molecular de 500.83. Regorafenib es prácticamente insoluble en agua, ligeramente soluble en acetonitrilo, metanol, etanol y acetato de etilo, y moderadamente soluble en acetona.
Los comprimidos de STIVARGA para administración oral están formulados como comprimidos de color rosa claro, de forma ovalada, con la palabra “BAYER” grabada en un lado y “40” en el otro. Cada comprimido contiene 40 mg de regorafenib en estado anhidro, que corresponde a 41.49 mg de regorafenib monohidrato, y los siguientes ingredientes inactivos: celulosa microcristalina, croscarmelosa sódica, estearato de magnesio, povidona y dióxido de silicio coloidal. El recubrimiento pelicular contiene los siguientes ingredientes inactivos: óxido férrico rojo, óxido férrico amarillo, lecitina (soja), polietilenglicol 3350, alcohol polivinílico, talco y dióxido de titanio.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Regorafenib es un inhibidor de moléculas pequeñas de múltiples cinasas unidas a membrana e intracelulares involucradas en funciones celulares normales y en procesos patológicos como la oncogénesis, angiogénesis tumoral, metástasis e inmunidad tumoral. En ensayos bioquímicos o celulares in vitro, regorafenib o sus principales metabolitos activos humanos M-2 y M-5 inhibieron la actividad de RET, VEGFR1, VEGFR2, VEGFR3, KIT, PDGFR-alfa, PDGFR-beta, FGFR1, FGFR2, TIE2, DDR2, TrkA, Eph2A, RAF-1, BRAF, BRAF V600E, SAPK2, PTK5, Abl y CSF1R a concentraciones de regorafenib que se han alcanzado clínicamente. En modelos in vivo, regorafenib demostró actividad antiangiogénica en un modelo de tumor de rata e inhibición del crecimiento tumoral en varios modelos de xenoinjerto de ratón, incluidos algunos de carcinoma colorrectal humano, estromal gastrointestinal y hepatocelular. Regorafenib también demostró actividad antimetastásica en un modelo de xenoinjerto de ratón y en dos modelos ortotópicos de ratón de carcinoma colorrectal humano.
12.2 Farmacodinámica
Electrofisiología cardíaca
Se evaluó el efecto de múltiples dosis de STIVARGA (160 mg una vez al día durante 21 días) en el intervalo QTc en un estudio abierto de un solo brazo en 25 pacientes con tumores sólidos avanzados. No se detectaron grandes cambios en el intervalo QTc medio (es decir, > 20 mseg) en el estudio.
12.3 Farmacocinética
Absorción
Después de una dosis única de 160 mg de STIVARGA en pacientes con tumores sólidos avanzados, regorafenib alcanza una concentración plasmática máxima media geométrica (Cmax) de 2,5 µg/ml en un tiempo medio de 4 horas y un área bajo la curva de concentración plasmática versus tiempo media geométrica (AUC) de 70,4 µg*h/ml. El AUC de regorafenib en estado estacionario aumenta menos que proporcionalmente con la dosis a dosis superiores a 60 mg. En estado estacionario, regorafenib alcanza una Cmax media geométrica de 3,9 µg/ml y un AUC medio geométrico de 58,3 µg*h/ml. El coeficiente de variación del AUC y Cmax está entre el 35 % y el 44 %.
La biodisponibilidad relativa media de los comprimidos en comparación con una solución oral es del 69 % al 83 %.
En un estudio del efecto de los alimentos, 24 hombres sanos recibieron una dosis única de 160 mg de STIVARGA en tres ocasiones distintas: en ayunas, con una comida rica en grasas y con una comida baja en grasas. Una comida rica en grasas (945 calorías y 54,6 g de grasa) aumentó el AUC medio de regorafenib en un 48 % y disminuyó el AUC medio de los metabolitos M-2 y M-5 en un 20 % y un 51 %, respectivamente, en comparación con el estado de ayuno. Una comida baja en grasas (319 calorías y 8,2 g de grasa) aumentó el AUC medio de regorafenib, M-2 y M-5 en un 36 %, 40 % y 23 %, respectivamente, en comparación con las condiciones de ayuno. STIVARGA se administró con una comida baja en grasas en los estudios CORRECT y GRID [ver Dosificación y administración (2.1), Estudios clínicos (14)].
Distribución
Regorafenib experimenta circulación enterohepática con múltiples picos de concentración plasmática observados durante el intervalo de dosificación de 24 horas. Regorafenib se une en gran medida (99,5 %) a las proteínas plasmáticas humanas.
Eliminación
Después de una dosis oral única de 160 mg de STIVARGA, las vidas medias de eliminación media geométrica (mínimo a máximo) de regorafenib y el metabolito M-2 en plasma son de 28 horas (14 a 58 horas) y 25 horas (14 a 32 horas), respectivamente. M-5 tiene una vida media de eliminación media (mínimo a máximo) más larga de 51 horas (32 a 70 horas).
Metabolismo
Regorafenib es metabolizado por CYP3A4 y UGT1A9. Los principales metabolitos circulantes de regorafenib medidos en estado estacionario en plasma humano son M-2 (N-óxido) y M-5 (N-óxido y N-desmetil). Ambos metabolitos tienen una actividad farmacológica in vitro y concentraciones en estado estacionario similares a las de regorafenib. M-2 y M-5 están altamente unidos a proteínas (99,8 % y 99,95 %, respectivamente).
Excreción
Aproximadamente el 71 % de una dosis radiomarcada se excretó en las heces (47 % como compuesto original, 24 % como metabolitos) y el 19 % de la dosis se excretó en la orina (17 % como glucurónidos) dentro de los 12 días posteriores a la administración de una solución oral radiomarcada a una dosis de 120 mg.
Poblaciones específicas
La edad, el sexo, la raza y el peso no tuvieron un efecto clínicamente significativo sobre la farmacocinética de regorafenib.
Insuficiencia hepática
Según un análisis farmacocinético poblacional, no se observaron diferencias clínicamente importantes en la exposición total media de regorafenib, incluidos M-2 y M-5, entre pacientes con función hepática normal (bilirrubina total y AST ≤ LSN, n=744), insuficiencia hepática leve (bilirrubina total ≤ LSN y AST > LSN o bilirrubina total > LSN a ≤1,5 veces el LSN, n=437) e insuficiencia hepática moderada (bilirrubina total >1,5 veces a ≤3 veces el LSN y cualquier AST, n=36). El análisis agrupado incluyó a 391 pacientes con CHC, de los cuales 116, 249 y 26 fueron categorizados como con función hepática normal, insuficiencia hepática leve y moderada, respectivamente. No se evaluó la farmacocinética de regorafenib en pacientes con insuficiencia hepática grave (bilirrubina total >3 veces el LSN).
Insuficiencia renal
Se evaluó la farmacocinética de regorafenib, M-2 y M-5 en 6 pacientes con insuficiencia renal grave (CLcr 15-29 ml/min) y 18 pacientes con función renal normal/leve (CLcr ≥60 ml/min) después de la administración de STIVARGA a una dosis de 160 mg al día durante 21 días. No se observaron diferencias en la exposición media en estado estacionario de regorafenib, M-2 o M-5 en pacientes con insuficiencia renal grave en comparación con pacientes con función renal normal. No se ha estudiado la farmacocinética de regorafenib en pacientes con enfermedad renal en etapa terminal en diálisis.
Estudios de interacción de medicamentos
Efecto de regorafenib sobre los sustratos del citocromo P450: estudios in vitro sugirieron que regorafenib es un inhibidor de CYP2C8, CYP2C9, CYP2B6, CYP3A4 y CYP2C19; M-2 es un inhibidor de CYP2C9, CYP2C8, CYP3A4 y CYP2D6, y M-5 es un inhibidor de CYP2C8. Estudios in vitro sugirieron que regorafenib no es un inductor de la actividad enzimática de CYP1A2, CYP2B6, CYP2C19 y CYP3A4.
Los pacientes con tumores sólidos avanzados recibieron dosis orales únicas de sustratos de CYP, 2 mg de midazolam (CYP3A4), 40 mg de omeprazol (CYP2C19) y 10 mg de warfarina (CYP2C9) o 4 mg de rosiglitazona (CYP2C8) una semana antes y dos semanas después de STIVARGA a una dosis de 160 mg una vez al día. No se observó ningún efecto clínicamente significativo en el AUC medio de rosiglitazona (N=12) o las concentraciones plasmáticas medias de omeprazol (N=11) medidas 6 horas después de la dosis o el AUC medio de midazolam (N=15). El AUC medio de warfarina (N=8) aumentó en un 25 % [ver Advertencias y precauciones (5.2)].
Efecto de los inductores fuertes del CYP3A4 sobre regorafenib: Veintidós hombres sanos recibieron una dosis única de 160 mg de STIVARGA solo y luego 7 días después de comenzar rifampicina. La rifampicina, un fuerte inductor del CYP3A4, se administró a una dosis de 600 mg diarios durante 9 días. El AUC medio de regorafenib disminuyó en un 50% y el AUC medio de M-5 aumentó en un 264%. No se observaron cambios en el AUC medio de M-2 [ver Interacciones farmacológicas (7.1)].
Efecto de los inhibidores fuertes del CYP3A4 sobre regorafenib: Dieciocho hombres sanos recibieron una dosis única de 160 mg de STIVARGA solo y luego 5 días después de comenzar ketoconazol. El ketoconazol, un fuerte inhibidor del CYP3A4, se administró a una dosis de 400 mg diarios durante 18 días. El AUC medio de regorafenib aumentó en un 33% y el AUC medio de M-2 y M-5 disminuyeron ambos en un 93% [ver Interacciones farmacológicas (7.2)].
Efecto de la neomicina sobre regorafenib: Veintisiete hombres sanos recibieron una dosis única de 160 mg de STIVARGA y luego 5 días después de comenzar neomicina. La neomicina, un antibiótico no absorbible, se administró a una dosis de 1 gramo tres veces al día durante 5 días. No se observó un efecto clínicamente significativo sobre el AUC medio de regorafenib; sin embargo, el AUC medio de M-2 disminuyó en un 76% y el AUC medio de M-5 disminuyó en un 86%. La disminución de la exposición de M-2 y M-5 puede resultar en una disminución de la eficacia de STIVARGA. No se han estudiado los efectos de otros antibióticos sobre la exposición de regorafenib y sus metabolitos activos.
Efecto de regorafenib sobre los sustratos de UGT1A1: Estudios in vitro mostraron que regorafenib, M-2 y M-5 inhiben competitivamente UGT1A9 y UGT1A1 a concentraciones terapéuticamente relevantes. Once pacientes recibieron quimioterapia de combinación que contenía irinotecán con STIVARGA a una dosis de 160 mg. El AUC medio de irinotecán aumentó en un 28% y el AUC medio de SN-38 aumentó en un 44% cuando se administró irinotecán 5 días después de la última de 7 dosis diarias de STIVARGA.
Efecto de regorafenib sobre los sustratos de BCRP: La administración de regorafenib (160 mg durante 14 días) antes de la administración de una dosis única de rosuvastatina (5 mg), un sustrato de BCRP, resultó en un aumento de 3,8 veces en la exposición media (AUC) de rosuvastatina y un aumento de 4,6 veces en la Cmax [ver Interacciones farmacológicas (7.3)].
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
No se han realizado estudios que examinen el potencial carcinogénico de regorafenib. El regorafenib en sí mismo no demostró genotoxicidad en ensayos in vitro o in vivo; sin embargo, un metabolito humano activo importante de regorafenib, (M-2), fue positivo para clastogenicidad, causando aberración cromosómica en células V79 de hámster chino.
No se han realizado estudios dedicados para examinar los efectos de regorafenib sobre la fertilidad; sin embargo, hubo hallazgos histológicos de atrofia tubular y degeneración en los testículos, atrofia en la vesícula seminal y restos celulares y oligospermia en los epidídimos en ratas macho a dosis similares a las de humanos a la dosis clínica recomendada según el AUC. En ratas hembra, hubo un aumento de los hallazgos de cuerpos lúteos necróticos en los ovarios a las mismas exposiciones. Hubo hallazgos similares en perros de ambos sexos en estudios de dosis repetidas a exposiciones de aproximadamente el 83% de la exposición humana a la dosis humana recomendada según el AUC. Estos hallazgos sugieren que regorafenib puede afectar negativamente la fertilidad en humanos.
13.2 Toxicología Animal y/o Farmacología
En un estudio crónico de dosis repetidas de 26 semanas en ratas, hubo un aumento dependiente de la dosis en el hallazgo de engrosamiento de la válvula atrioventricular. A una dosis que resultó en una exposición de aproximadamente el 12% de la exposición humana a la dosis recomendada, este hallazgo estuvo presente en la mitad de los animales examinados.
14 ESTUDIOS CLÍNICOS
14.1 Cáncer Colorrectal
La eficacia clínica y seguridad de STIVARGA fueron evaluadas en un ensayo internacional, multicéntrico, aleatorizado (2:1), doble ciego, controlado con placebo [Estudio “Pacientes con cáncer COloRectal metastásico tratados con REgorafenib o plaCebo después del fracaso de la Terapia estándar” (CORRECT); NCT 01103323)] en 760 pacientes con cáncer colorrectal metastásico previamente tratado. La principal medida de eficacia fue la supervivencia global (OS); las medidas adicionales de eficacia incluyeron la supervivencia libre de progresión (PFS) y la tasa de respuesta tumoral global.
Los pacientes fueron aleatorizados para recibir 160 mg de regorafenib por vía oral una vez al día (N=505) más el mejor cuidado de soporte (BSC) o placebo (N=255) más BSC durante los primeros 21 días de cada ciclo de 28 días. STIVARGA se administró con un desayuno bajo en grasas que contiene menos del 30% de grasa [ver Dosificación y Administración (2.1), Farmacología Clínica (12.3)]. El tratamiento continuó hasta la progresión de la enfermedad o toxicidad inaceptable.
Los datos demográficos basales fueron: mediana de edad 61 años, 61% hombres, 78% blancos, y todos los pacientes tenían un estado funcional ECOG de 0 o 1. Los sitios primarios de la enfermedad fueron colon (65%), recto (29%), o ambos (6%). Se reportó historial de evaluación de KRAS para 729 (96%) pacientes; se reportó que 430 (59%) de estos pacientes tenían mutación de KRAS. La mediana del número de líneas previas de terapia para la enfermedad metastásica fue 3. Todos los pacientes recibieron tratamiento previo con quimioterapia basada en fluoropirimidina, oxaliplatino e irinotecán, y con bevacizumab. Todos excepto un paciente con tumores KRAS mutación-negativos recibieron panitumumab o cetuximab.
La adición de STIVARGA a BSC resultó en una mejora estadísticamente significativa en la supervivencia en comparación con placebo más BSC (ver Tabla 7 y Figura 1).
|
STIVARGA (N=505) |
Placebo (N=255) |
|
|
Supervivencia Global |
||
|
Número de Muertes (%) |
275 (55%) |
157 (62%) |
|
6.4 |
5.0 |
|
(5.8, 7.3) |
(4.4, 5.8) |
|
0.77 (0.64, 0.94) |
|
|
0.0102 |
|
|
Supervivencia Libre de Progresión |
||
|
Número de Muertes o Progresiones (%) |
417 (83%) |
231 (91%) |
|
2.0 |
1.7 |
|
(1.9, 2.3) |
(1.7, 1.8) |
|
0.49 (0.42, 0.58) |
|
|
<0.0001 |
|
|
Tasa de Respuesta Global |
||
|
5 (1%) |
1 (0.4%) |
|
0.3%, 2.3% |
0%, 2.2% |
- a CI=intervalo de confianza.
- bEstratificado por región geográfica y tiempo desde el diagnóstico de enfermedad metastásica.
- c Cruzó el límite de O’Brien-Fleming (valor p bilateral < 0.018) en el segundo análisis intermedio.
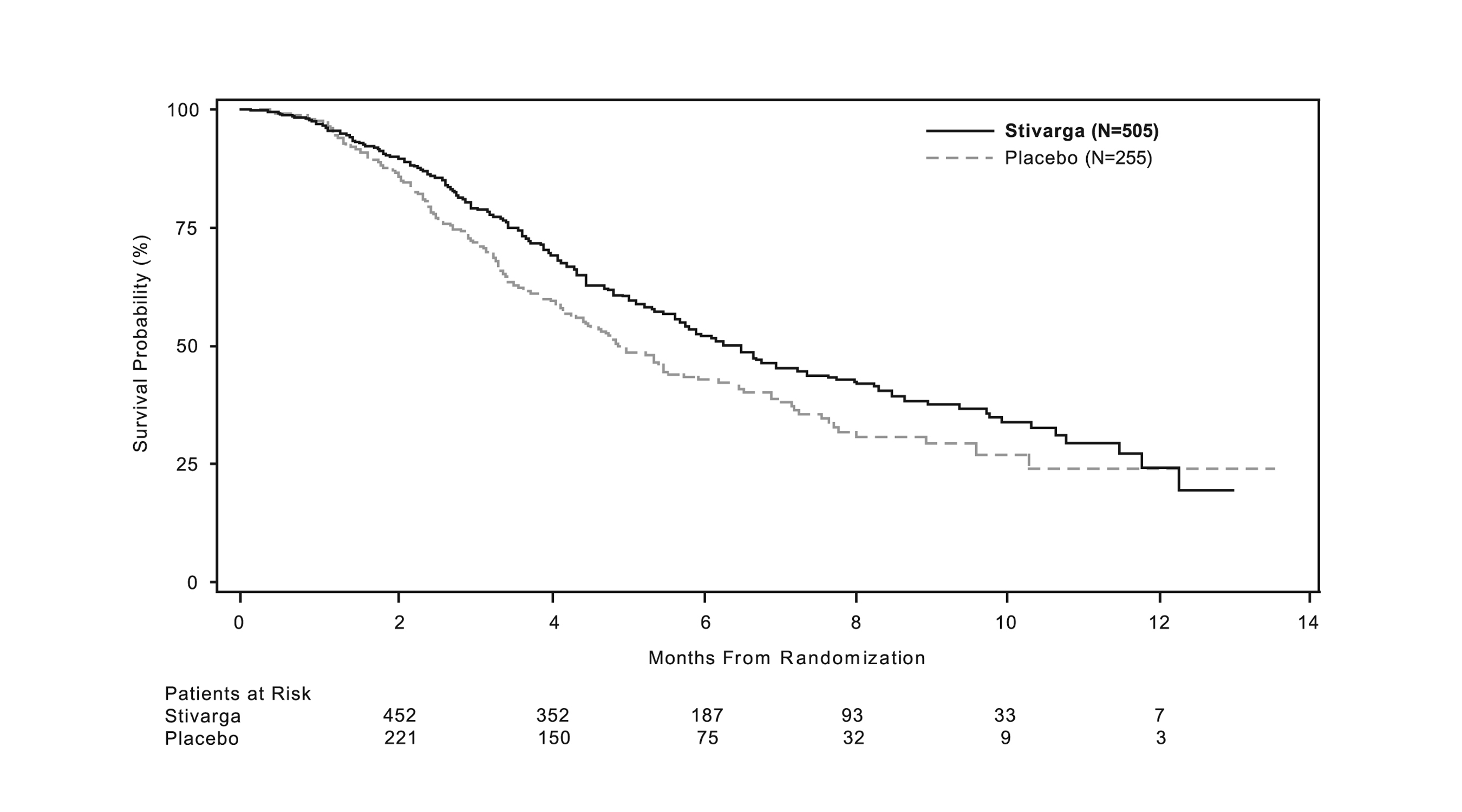
Figura 1: Curvas de Kaplan-Meier de Supervivencia Global
14.2 Tumores del estroma gastrointestinal
La eficacia y seguridad de STIVARGA se evaluaron en un ensayo internacional, multicéntrico, aleatorizado (2:1), doble ciego, controlado con placebo [Estudio “GIST Regorafenib In progressive Disease” (GRID); NCT 01271712] en pacientes con tumor del estroma gastrointestinal (GIST) irresecable, localmente avanzado o metastásico, que habían sido previamente tratados con mesilato de imatinib y malato de sunitinib. La aleatorización se estratificó por línea de terapia (tercera vs. cuarta o más) y región geográfica (Asia vs. resto del mundo).
La principal medida de eficacia de GRID fue la supervivencia libre de progresión (SLP) según la evaluación de la enfermedad por revisión radiológica independiente utilizando los criterios RECIST 1.1 modificados, en los que los ganglios linfáticos y las lesiones óseas no eran lesiones diana y el nuevo nódulo tumoral de crecimiento progresivo dentro de una masa tumoral preexistente era progresión. La medida de resultado secundaria clave fue la supervivencia global.
Los pacientes fueron aleatorizados para recibir 160 mg de regorafenib por vía oral una vez al día (N=133) más el mejor cuidado de soporte (BSC) o placebo (N=66) más BSC durante los primeros 21 días de cada ciclo de 28 días. El tratamiento continuó hasta la progresión de la enfermedad o una toxicidad inaceptable. En GRID, la mediana de edad de los pacientes fue de 60 años, el 64% eran hombres, el 68% eran blancos y todos los pacientes tenían un estado de rendimiento ECOG basal de 0 (55%) o 1 (45%). En el momento de la progresión de la enfermedad según la evaluación de la revisión central, se rompió el ciego del estudio y a todos los pacientes se les ofreció la oportunidad de tomar STIVARGA a discreción del investigador.
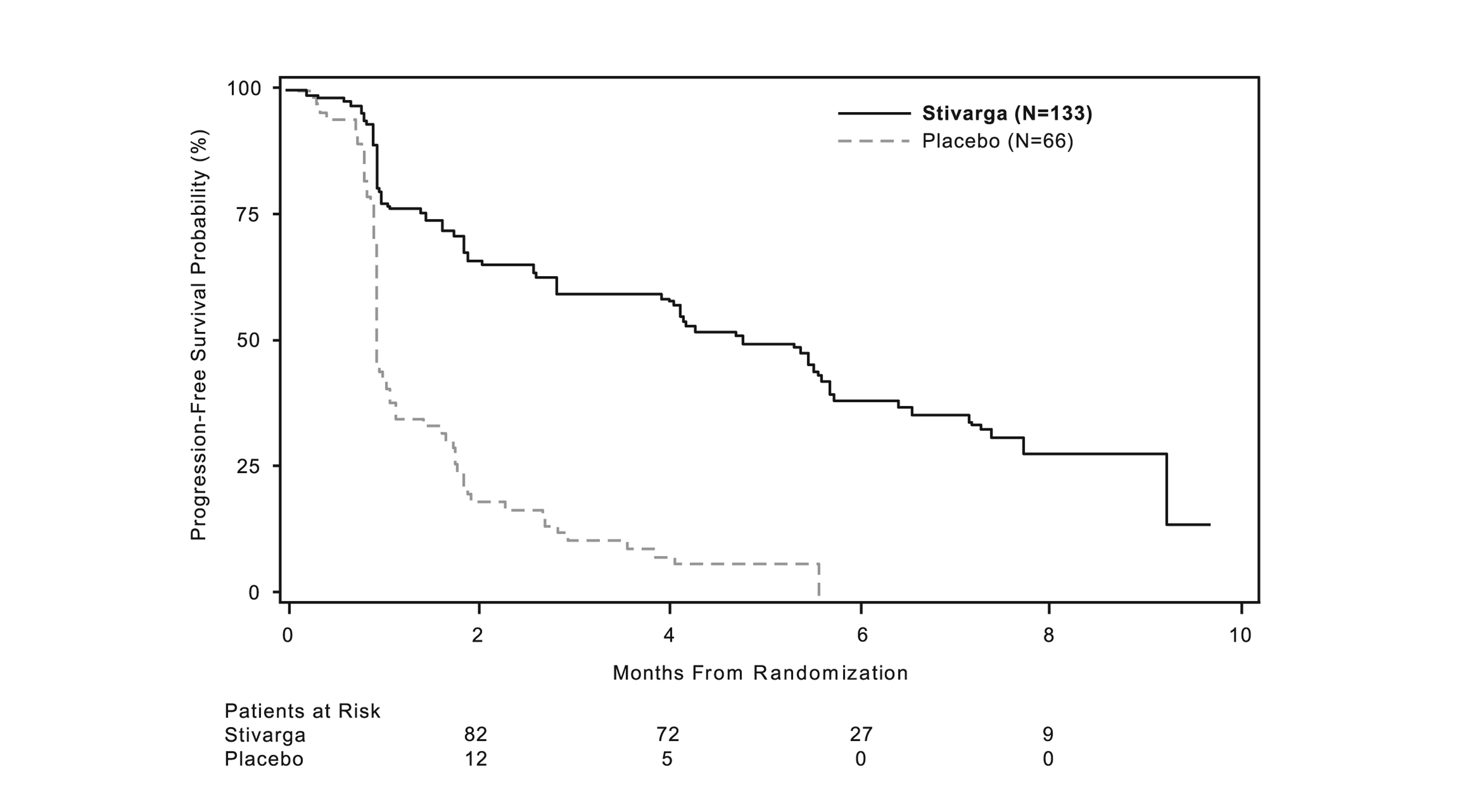
Se demostró una mejora estadísticamente significativa en la SLP entre los pacientes tratados con STIVARGA en comparación con el placebo (ver Tabla 8 y Figura 2).
No hubo diferencia estadísticamente significativa en la supervivencia global en el análisis final de SG, realizado a los 162 eventos de SG (Tabla 8). El 58% (88%) de los pacientes tratados con placebo cruzaron a STIVARGA abierto después de la progresión de la enfermedad.
|
STIVARGA (N=133) |
Placebo (N=66) |
|
|
Supervivencia Libre de Progresión |
||
|
Número de Muertes o Progresiones (%) |
82 (62%) |
63 (96%) |
|
SLP media en meses (IC 95%) |
4.8 (3.9, 5.7) |
0.9 (0.9, 1.1) |
|
HR (IC 95%) |
0.27 (0.19, 0.39) |
|
|
valor pa |
<0.0001 |
|
|
Supervivencia Global |
||
|
Número de Muertes (%) |
109 (82%) |
53 (80.3%) |
|
SG media en meses (IC 95%) |
17.4 (14.9, 20.2) |
17.4 (12.3, 21.0) |
|
HR (IC 95%) |
0.91 (0.65, 1.27) |
|
|
valor pa |
0.5716 |
|
a Valor p de 2 colas por prueba de log-rank estratificada por línea de tratamiento y región geográfica.
14.3 Carcinoma Hepatocelular (CHC)
La eficacia clínica y seguridad de STIVARGA se evaluaron en un ensayo internacional, multicéntrico, aleatorizado (2:1), doble ciego, controlado con placebo [Estudio “REgorafenib after SORafenib in patients with hepatoCEllular carcinoma” (RESORCE); NCT 01774344]. El estudio enroló adultos con carcinoma hepatocelular Child-Pugh A y Categoría B o C del Barcelona Clinic Liver Cancer Stage, con progresión documentada de la enfermedad después de sorafenib. La duración media del tratamiento previo con sorafenib fue de 7,8 meses; los pacientes que suspendieron permanentemente sorafenib debido a toxicidad o que no pudieron tolerar dosis de sorafenib de 400 mg una vez al día no fueron elegibles.
Los pacientes fueron aleatorizados para recibir 160 mg de regorafenib por vía oral una vez al día más el mejor cuidado de soporte (BSC) o placebo equivalente más BSC durante los primeros 21 días de cada ciclo de 28 días hasta la progresión de la enfermedad o una toxicidad inaceptable. La aleatorización se estratificó por región geográfica (Asia vs resto del mundo), estado funcional ECOG (0 vs 1), niveles de alfa-fetoproteína (<400 ng/mL vs ≥400 ng/mL), enfermedad extrahepática (presencia vs ausencia) e invasión macrovascular (presencia vs ausencia). La principal medida de eficacia fue la supervivencia global (SG). Las medidas de resultado adicionales fueron la supervivencia libre de progresión (SLP), la tasa de respuesta tumoral global (TRG) y la duración de la respuesta según la evaluación de los investigadores mediante RECIST 1.1 y mediante RECIST modificado (mRECIST) para CHC. Los pacientes continuaron la terapia con STIVARGA hasta la progresión clínica o radiológica de la enfermedad o una toxicidad inaceptable.
Las características de la población del estudio fueron una mediana de edad de 63 años (rango de 19 a 85 años); 88% hombres; 41% asiáticos, 36% blancos y 21% no reportados; 66% tenían un estado funcional ECOG (PS) de 0 y 34% tenían un PS ECOG de 1; 98% tenían Child-Pugh A y 2% tenían Child-Pugh B. Los factores de riesgo para la cirrosis subyacente incluyeron hepatitis B (38%), consumo de alcohol (25%), hepatitis C (21%) y hepatitis esteatósica no alcohólica (7%). La invasión vascular macroscópica o la diseminación tumoral extrahepática estuvo presente en el 81% de los pacientes. El estadio del Barcelona Clinic Liver Cancer (BCLC) fue C en el 87% y B en el 13% de los pacientes. Todos los pacientes recibieron sorafenib previamente y el 61% recibió procedimientos previos de embolización transarterial o quimioinfusión locorregional.
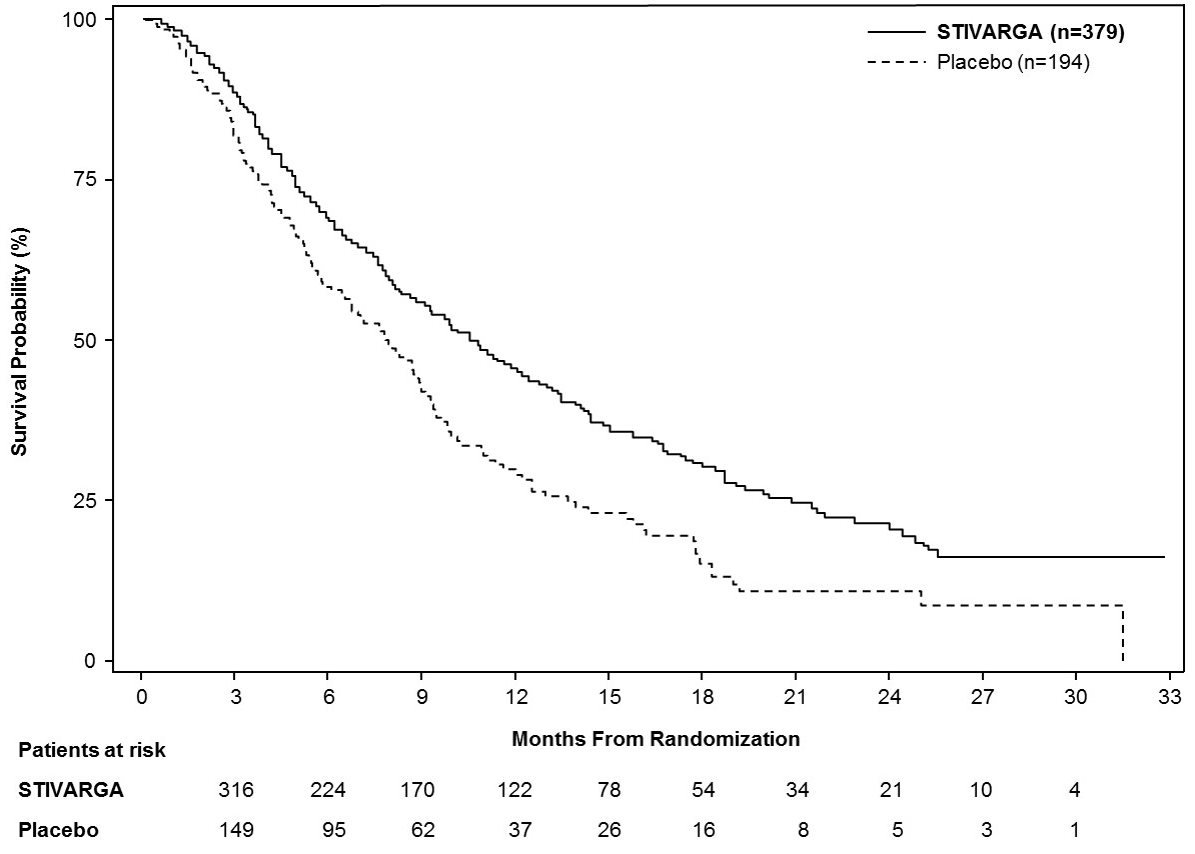
Los resultados de eficacia se resumen en la Tabla 9 y la Figura 3 a continuación.
|
|
STIVARGA n=379 |
Placebo n=194 |
|
Supervivencia global |
||
|
Número de muertes (%) |
233 (62) |
140 (72) |
|
Mediana de SG en meses (IC del 95%a) |
10.6 (9.1, 12.1) |
7.8 (6.3, 8.8) |
|
Hazard Ratiob (IC del 95%a) |
0.63 (0.50, 0.79) |
|
|
Valor pc |
<0.0001 |
|
|
Supervivencia libre de progresión (mRECIST) |
||
|
Número de eventos (%) |
293 (77) |
181(93) |
|
Enfermedad progresiva |
274 (72) |
173 (89) |
|
Muerte |
19 (5) |
8 (4) |
|
Mediana de SLP en meses (IC del 95%a) |
3.1 (2.8, 4.2) |
1.5 (1.4, 1.6) |
|
Hazard Ratiob (IC del 95%a) |
0.46 (0.37, 0.56) |
|
|
Valor pc |
<0.0001 |
|
|
Supervivencia libre de progresión (RECIST 1.1) |
||
|
Número de eventos (%) |
288 (76) |
184 (95) |
|
Enfermedad progresiva |
270 (71) |
175 (90) |
|
Muerte |
18 (5) |
9 (5) |
|
Mediana de SLP en meses (IC del 95%a) |
3.4 (2.9, 4.2) |
1.5 (1.4, 1.5) |
|
Hazard Ratiob (IC del 95%a) |
0.43 ( 0.35, 0.52) |
|
|
Respuesta general (mRECIST) |
||
|
Tasa de respuesta general |
11% |
4% |
|
IC del 95%a |
(8%, 14%) |
(2%, 8%) |
|
Respuesta completa |
0.5% |
0 |
|
Respuesta parcial |
10% |
4% |
|
Respuesta General (RECIST 1.1) |
||
|
Tasa de Respuesta General |
7% |
3% |
|
IC del 95%a |
(4%, 10%) |
(1%, 6%) |
|
Respuesta Completa |
0 |
0 |
|
Respuesta Parcial |
7% |
3% |
- aIC=intervalo de confianza.
- bEstimado con el modelo de riesgos proporcionales de Cox estratificado por región geográfica, estado funcional ECOG, nivel de alfa-fetoproteína, presencia versus ausencia de enfermedad extrahepática y presencia versus ausencia de invasión macrovascular.
- cPrueba de log-rank estratificada por región geográfica, estado funcional ECOG, nivel de alfa-fetoproteína, presencia versus ausencia de enfermedad extrahepática y presencia versus ausencia de invasión macrovascular.
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Forma de Suministro
Los comprimidos se suministran en lo siguiente:
Paquetes que contienen tres frascos, cada frasco contiene 28 comprimidos, para un total de 84 comprimidos por paquete
(NDC 50419-171-03).
Paquetes que contienen cuatro frascos, cada frasco contiene 21 comprimidos, para un total de 84 comprimidos por paquete
(NDC 50419-171-06).
Almacenamiento y Manipulación
Almacene STIVARGA a 25°C (77°F); se permiten excursiones de 15 a 30°C (59 a 86°F) [ver USP Controlled Room Temperature].
Almacene los comprimidos en el frasco original y no retire el desecante. Mantenga el frasco bien cerrado después de abrirlo por primera vez.
Deseche los comprimidos no utilizados 7 semanas después de abrir el frasco. Elimine los comprimidos no utilizados de acuerdo con los requisitos locales.
17 INFORMACIÓN DE ASESORAMIENTO AL PACIENTE
Aconseje al paciente que lea la información para el paciente aprobada por la FDA (Información para el paciente).
Hepatotoxicidad
Informe a los pacientes que deberán someterse a un monitoreo del daño hepático y que deben informar de inmediato a su proveedor de atención médica cualquier signo o síntoma de daño hepático grave [ver Advertencias y precauciones (5.1), Uso en poblaciones específicas (8.6)].
Infecciones
Aconseje a los pacientes que se comuniquen con su proveedor de atención médica si experimentan signos y síntomas de infección [ver Advertencias y precauciones (5.2)].
Hemorragia
Aconseje a los pacientes que se comuniquen con su proveedor de atención médica por sangrado inusual, hematomas o síntomas de sangrado, como mareos [ver Advertencias y precauciones (5.3)].
Perforación gastrointestinal o fístula
Aconseje a los pacientes que se comuniquen de inmediato con un proveedor de atención médica si experimentan dolor abdominal intenso, hinchazón persistente del abdomen, fiebre alta, escalofríos, náuseas, vómitos o deshidratación [ver Advertencias y precauciones (5.4)].
Toxicidad dermatológica
Aconseje a los pacientes que se comuniquen con su proveedor de atención médica si experimentan cambios en la piel, incluidos HFSR, erupción cutánea, dolor, ampollas, sangrado o hinchazón [ver Advertencias y precauciones (5.5)].
Hipertensión
Informe a los pacientes que deberán someterse a un monitoreo de la presión arterial y que deben comunicarse con su proveedor de atención médica si la presión arterial está elevada o si se presentan síntomas de hipertensión, incluidos dolor de cabeza intenso, mareos o síntomas neurológicos [ver Advertencias y precauciones (5.6)].
Isquemia e infarto cardíacos
Aconseje a los pacientes que busquen ayuda de emergencia inmediata si experimentan dolor en el pecho, dificultad para respirar, mareos o sensación de desmayo [ver Advertencias y precauciones (5.7)].
Síndrome de leucoencefalopatía posterior reversible
Aconseje a los pacientes que se comuniquen con su proveedor de atención médica si experimentan signos y síntomas de RPLS [ver Advertencias y precauciones (5.8)].
Riesgo de deterioro de la cicatrización de heridas
Informe a los pacientes que STIVARGA puede alterar la cicatrización de las heridas. Informe a los pacientes que se recomienda la interrupción temporal de STIVARGA antes de cualquier cirugía electiva [ver Advertencias y precauciones (5.9)].
Toxicidad embriofetal
Informe a los pacientes que regorafenib puede causar daño fetal. Informe a las mujeres embarazadas sobre el riesgo potencial para el feto [ver Advertencias y precauciones (5.10), Uso en poblaciones específicas (8.1, 8.3)].
Mujeres y hombres con capacidad reproductiva
- •
- Informe a las mujeres con potencial reproductivo sobre la necesidad de usar un método anticonceptivo eficaz durante el tratamiento con STIVARGA y durante 2 meses después de finalizar el tratamiento. Indique a las mujeres con potencial reproductivo que se comuniquen de inmediato con su proveedor de atención médica si se sospecha o confirma un embarazo durante o dentro de los 2 meses posteriores a la finalización del tratamiento con STIVARGA [ver Advertencias y precauciones (5.10) y Uso en poblaciones específicas (8.1, 8.3)].
- •
- Informe a los hombres con capacidad reproductiva sobre la necesidad de usar un método anticonceptivo eficaz durante el tratamiento con STIVARGA y durante 2 meses después de finalizar el tratamiento [ver Uso en poblaciones específicas (8.3)].
Lactancia
Informe a las madres lactantes que se desconoce si regorafenib está presente en la leche materna y analice si se debe suspender la lactancia o suspender regorafenib [ver Uso en poblaciones específicas (8.2)].
Administración
- •
- Aconseje a los pacientes que traguen la tableta de STIVARGA entera con agua a la misma hora todos los días después de una comida baja en grasas. Informe a los pacientes que la comida baja en grasas debe contener menos de 600 calorías y menos del 30 % de grasas [ver Dosis y administración (2.1)].
- •
- Aconseje a los pacientes que guarden el medicamento en el envase original. No coloque el medicamento en pastilleros diarios o semanales. Deseche las tabletas restantes 7 semanas después de abrir el frasco. Cierre bien el frasco después de cada apertura y mantenga el desecante en el frasco [ver Cómo se suministra (16)].
Instrucciones de dosificación
Aconseje a los pacientes que tomen STIVARGA después de una comida baja en grasas. Aconseje a los pacientes que tomen cualquier dosis olvidada el mismo día, tan pronto como lo recuerden, y que no deben tomar dos dosis el mismo día para compensar una dosis olvidada el día anterior [ver Dosis y administración (2.1)].
Fabricado para:
Bayer HealthCare Pharmaceuticals Inc.
Whippany, NJ 07981 USA
Prospecto para el paciente
|
Información para el paciente STIVARGA (sti-VAR-gah) (regorafenib) tabletas |
|
|
¿Cuál es la información más importante que debo saber sobre STIVARGA? STIVARGA puede causar efectos secundarios graves, que incluyen: Problemas hepáticos. STIVARGA puede causar problemas hepáticos que pueden ser graves y, a veces, provocar la muerte. Su proveedor de atención médica le hará análisis de sangre para verificar la función hepática antes de comenzar a tomar STIVARGA y durante su tratamiento con STIVARGA para detectar problemas hepáticos. Informe a su proveedor de atención médica de inmediato si presenta alguno de estos síntomas de problemas hepáticos durante el tratamiento: |
|
|
|
|
¿Qué es STIVARGA? STIVARGA es un medicamento recetado que se usa para tratar a personas con:
No se sabe si STIVARGA es seguro y efectivo en niños menores de 18 años. |
|
|
Antes de tomar STIVARGA, informe a su proveedor de atención médica sobre todas sus afecciones médicas, incluso si:
Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos a base de hierbas. STIVARGA puede afectar la forma en que funcionan otros medicamentos, y otros medicamentos pueden afectar cómo funciona STIVARGA. |
|
|
¿Cómo debo tomar STIVARGA?
Si toma demasiado STIVARGA, llame a su proveedor de atención médica o acuda a la sala de emergencias más cercana de inmediato. |
|
|
¿Qué debo evitar mientras tomo STIVARGA? Evite beber jugo de toronja y tomar hierba de San Juan durante el tratamiento con STIVARGA. Estos pueden afectar la forma en que funciona STIVARGA. |
|
|
¿Cuáles son los posibles efectos secundarios de STIVARGA? STIVARGA puede causar efectos secundarios graves, que incluyen:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Los efectos secundarios más comunes de STIVARGA incluyen: |
|
|
|
|
Su proveedor de atención médica puede cambiar su dosis, suspender temporalmente o suspender permanentemente el tratamiento con STIVARGA si tiene ciertos efectos secundarios. Estos no son todos los posibles efectos secundarios de STIVARGA. Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088. |
|
|
¿Cómo almaceno STIVARGA?
Mantenga STIVARGA y todos los medicamentos fuera del alcance de los niños. |
|
|
Información general sobre el uso seguro y eficaz de STIVARGA. Los medicamentos a veces se recetan para fines distintos a los enumerados en un folleto de Información para el paciente. No use STIVARGA para una afección para la cual no fue recetado. No le dé STIVARGA a otras personas, incluso si tienen los mismos síntomas que usted. Puede hacerles daño. Puede solicitar a su proveedor de atención médica o farmacéutico información sobre STIVARGA que está escrita para profesionales de la salud. |
|
|
¿Cuáles son los ingredientes de STIVARGA? Ingrediente activo: regorafenib Ingredientes inactivos: celulosa microcristalina, croscarmelosa sódica, estearato de magnesio, povidona y dióxido de silicio coloidal. Recubrimiento: óxido de hierro rojo, óxido de hierro amarillo, lecitina (soja), polietilenglicol 3350, alcohol polivinílico, talco y dióxido de titanio. Fabricado para Bayer HealthCare Pharmaceuticals Inc., Whippany, NJ 07981 USA. © 2017 Bayer HealthCare Pharmaceuticals Inc. |
|
- Esta Información para el Paciente ha sido aprobada por la Administración de Alimentos y Medicamentos de los Estados Unidos. Revisado: 02/2020
PANEL DE VISUALIZACIÓN PRINCIPAL DEL PAQUETE/ETIQUETA
NDC 50419-171-03
Rx Only
Stivarga
(regorafenib) comprimidos
40 mg
3 x 28 comprimidos
para administración oral
